Introduction
Holoprosencephaly (HPE) is a rare congenital brain abnormality of the midline, presenting in three forms: alobar, semilobar, and lobar. It results from a defect in the cleavage of the prosencephalon into cerebral hemispheres [1]. It is a rare fetal pathology with significant etiological heterogeneity. It affects one in 15,000 to 16,000 births [2].This pathology has an extremely poor fetal prognosis, particularly for the alobar form [2]. Bearing this in mind, we report the case of an alobar HPE diagnosed during a routine ultrasound examination and aim to raise awareness on the importance of early prenatal detection and counselling.
Case report
A 37-year-old patient with a medical history of poorly controlled type 2 diabetes under insulin treatment (recent HbA1c at 8.2%), and an infection with SARS-CoV-2 at 17 weeks of gestation during this pregnancy was referred to us at 21 weeks gestation following the discovery at the second-trimester ultrasound of an alobar HPE.
The patient had five gestations, two cesarean sections, non-consanguineous marriage, a history of two unexplored terminated pregnancies, a cesarean delivery at term with a healthy baby and a history of medical termination of pregnancy at 20 weeks for anencephaly via cesarean section which was not further investigated at the mother's request.
The current pregnancy was not regularly monitored by the patient, and the first-trimester scan was not performed. Folic acid supplementation was initiated at 7 weeks.
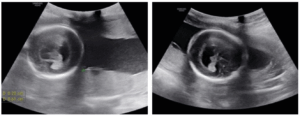
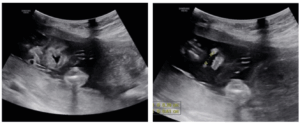
Ultrasound performed when referred shows a significant dilation of a single ventricular cavity, associated with fusion of the thalami, absence of the interhemispheric fissure, and agenesis of the corpus callosum (Figure 1) associated with a cleft lip and palate (Figure 2).
A multidisciplinary meeting including obstetricians, neonatologists and embryologists concluded that termination of pregnancy was necessary given the poor neurological prognosis. A psychological accompaniment was provided to the couple, helping to avoid psychological and emotional trauma for the parents, and genetic counseling was also offered.
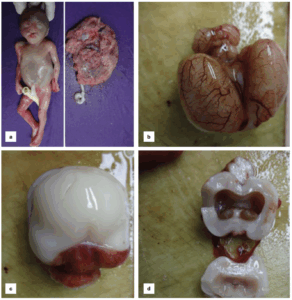
Pathologic examination of the fetus revealed alobar HPE with a premaxillary agenesis (Figure 3).
A conventional fetal karyotype denied any chromosomal abnormalities (46, XX). Samples of the placenta, amniotic fluid, and umbilical cord blood were PCR tested for PCR SARS-CoV-2 virus, which were negative.
Discussion
HPE, although rare, is the most common malformation of the brain and face. It results from a cleavage abnormality of the prosencephalon and is characterized by the formation of a median hemispheric mass between days 18 and 28 of pregnancy [1,3]. It affects 1 in 10,000 live births occurring more in females than males [4,5].
During primary neurulation, the neural tube closes and gives rise to three primary brain vesicles: the prosencephalon, mesencephalon, and rhombencephalon. In the following ventral induction phase, which takes place between the fifth and tenth weeks of gestation, these vesicles undergo expansion and subdivision into secondary vesicles. Specifically, the prosencephalon differentiates into the telencephalon and diencephalon. Disruptions in critical signaling pathways, such as Sonic hedgehog (Shh), can result in HPE [6].
HPE is classified into two categories based on the extent of cerebral hemisphere differentiation. The first category, defined by DeMyer and Zeman, includes the classic forms arranged by severity: alobar, semilobar, and lobar. The second category refers to the median interhemispheric variant, also called syntelencephaly [6].
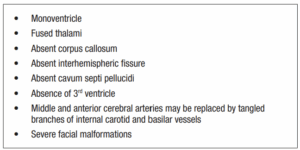
Alobar HPE, the most severe form, affects approximately two-thirds of individuals [7] and is characterized by a single ventricle, fused thalami, absence of the corpus callosum and a severe facial dysmorphism [3,8-10].
Ultrasound abnormalities found in alobar HPE are presented in Table 1. (Table 1)
HPEs are characterized by a high degree of etiological heterogeneity with most cases being sporadic. However, chromosomal abnormalities are found in 25% to 50% of affected individuals. Trisomy 13 accounts for 75% of these cases, followed by triploidy (20%) and trisomy 18 (1% to 2%). Additionally, single-gene disorders have also been reported [4,7,8,11,12]. However, environmental factors such as low socioeconomic status, maternal diabetes, alcohol consumption, retinoid use, and certain congenital infections, such as cytomegalovirus, also contribute to HPE risk [1,6,13,14].
In our case, the antenatal ultrasound scan successfully detected the key findings of alobar HPE. These findings included a single ventricular cavity, associated with fusion of the thalami, absence of the interhemispheric fissure, and agenesis of the corpus callosum associated with a cleft lip and palate. Chromosomal abnormalities were not found in our case, nor was infection a likely factor in the outcome of this pregnancy; however, type 2 diabetes may be associated with this pathology.
Alobar HPE is incompatible with life. This form, the most severe, confirmed in postnatally is often associated with a high mortality rate. Approximately 33% of affected newborns die within the first 24 hours, and 58% die within the first month [6]. Hence, early and thorough sonography may provide a means to the early intervention by proper counselling for possible termination of the pregnancy {8,15]. The antenatal diagnosis of HPE is based on conventional obstetrical ultrasound completed by fetal magnetic resonance imaging (MRI). Ultrasound diagnosis of HPE can be made early in the first trimester of pregnancy, allowing early termination of the pregnancy, particularly for the alobar and semi-lobar forms [13,16]. A MRI is used for diagnosis in the third trimester [17].
On the MRI, the basic structure of the cerebral hemispheres is lost, with variable amounts of residual cortex. Features include: single midline monoventricle (or holosphere), absent midline structures, dorsal cyst of HPE, absent, fused or normal optic nerves and middle and anterior cerebral arteries may be replaced by tangled branches of internal carotid and basilar vessels [9].
Associated craniofacial features may also be present which include: proboscis, mono-orbit/cyclopia, mononostril, hypotelorism, cebocephaly [9]. This highlights the importance of prenatal monitoring, which allows for early diagnosis and appropriate management. According to the International Society of Ultrasound in Obstetrics and Gynecology, the chances of HPE recurring can differ significantly from one family to another. Thus, genetic counseling offers tailored risk evaluations and insights, taking into account the family’s medical history, possible genetic factors, and any relevant environmental influences [10].
Conclusion
HPE is a rare congenital brain malformation. The diagnosis can be made through prenatal ultrasound by detecting a dilation of a single ventricular cavity and the absence of the interhemispheric fissure. Sonographic abnormalities of the face can also be detected. Chromosomal abnormalities, genetic factors, poorly controlled diabetes, and maternal-fetal infections are the most implicated causes, highlighting the importance of genetic counseling for parents and proper prenatal care. Termination of pregnancy is necessary as this neurological abnormality is fatal.