Introduction
Primary angiosarcoma of the breast (PAB) is an extremely rare malignant tumor [1]. Its aggressive behavior, coupled with the very limited number of published cases, means that the condition remains poorly characterized, and evidence-based management guidelines are still lacking. Because of this scarcity, clinicians may not immediately consider PAB when evaluating breast lesions, contributing to delayed diagnosis and suboptimal initial management. This case report describes the clinical course of a 27-year-old woman diagnosed with PAB. By detailing her presentation, evaluation, treatment, and early outcomes, we aim to provide practical insights that may support clinicians facing similar scenarios. In addition, this report includes a brief review of current literature, underscoring the importance of maintaining a high index of suspicion for this rare entity. The manuscript has been prepared in accordance with CARE guidelines.
Case report
We report the case of a 27-year-old Caucasian woman who detected a painful, warm mass in the right internal subareolar region. She had no significant medical, surgical, or gynecological history. Her mother had been diagnosed with breast cancer at 58, with no known hereditary mutation. On clinical examination, the patient presented an indurated, poorly defined mass, without axillary lymphadenopathy.
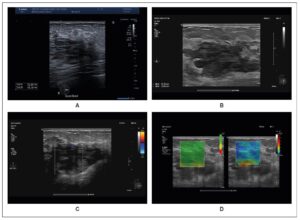
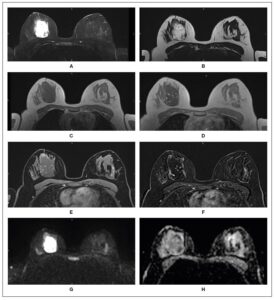
Initial ultrasonography revealed a heterogeneous 3.5 × 2 cm mass, and cytology suggested a benign hemorrhagic–inflammatory lesion (Figure 1). After a course of antibiotics, follow-up MRI demonstrated a well-demarcated 6 × 4 × 4.5 cm mass with T2 hyperintensity and diffusion, features compatible with an abscess (Figure 2). The lesion also showed progressive, atypical contrast enhancement, prompting further sampling. Microbiopsies revealed benign granulation tissue undergoing fibro-hyaline organization with telangiectatic vascular clefts (Nottingham Class B2).
Three months later, the lesion had increased in size. Ultrasound was used as the initial imaging modality due to its accessibility. It demonstrated a multilobulated, poorly defined 5 × 4 × 2 cm mass, heterogeneous, compressible, and non-indurated on elastography, with no detectable vascularization on color Doppler (Figure 1). Fine-needle aspiration—performed first due to the hemorrhagic risk associated with suspected vascularity—yielded only blood, and cytology remained nondiagnostic (C1 Nottingham). A vascular tumor was therefore considered.
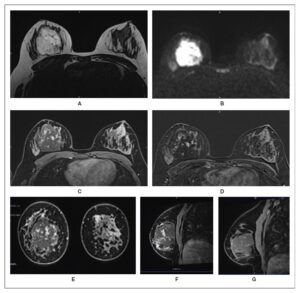
Repeat breast MRI showed that the lesion had grown to 6 × 5 × 6 cm (Figure 3). It appeared well-defined on T2-weighted imaging, with marked diffusion hypersignal and no axillary lymphadenopathy. Angiosarcoma became a primary diagnostic consideration. A second biopsy confirmed the diagnosis of a well-differentiated, low-grade primary angiosarcoma of the breast (Figure 4). Thoraco-abdominal CT and PET-CT revealed no distant metastases, and the lesion showed no FDG uptake on PET-CT.
Treatment consisted of a simple right mastectomy without lymph node dissection, including resection of the fascia and the medial portion of the pectoral muscle to achieve a 3-cm clear margin. Final histopathology confirmed a 5.5-cm low-grade primary angiosarcoma with negative margins and no c-myc amplification. Comprehensive genetic analysis identified no somatic mutations.
The patient underwent breast reconstruction using a large dorsal flap. At one year, clinical and radiologic follow-up remained reassuring, with no signs of recurrence. She continues close surveillance every six months with both gynecology and oncology teams. A PET-CT scan and breast MRI are scheduled yearly.
Discussion
This case report highlights the challenges involved in diagnosing and treating PAB. In young women, a painful breast mass is rarely initially suspected to be PAB, particularly when MRI findings suggest a poorly vascularized lesion, which may be misinterpreted as a breast abscess. However, lack of response to antibiotic therapy should prompt timely reassessment of the diagnosis. In this case, diagnostic uncertainty was further compounded by discordance between imaging findings and biopsy results, likely related to the low-grade nature of the tumor. Histologically, this tumor is characterized by anastomosing, irregular, non-atypical vascular channels that infiltrate the surrounding breast tissue [1,2]. On dynamic MRI, low-flow vascular channels may appear as a “blood-filled sponge,” accounting for the marked T2 hyperintensity observed, rather than representing purulent necrosis as initially interpreted.
PAB is a rare and frequently misdiagnosed aggressive vasoforming mesenchymal angiosarcoma that only rarely arises as a primary tumor of the breast [1,3]. It carries a poor prognosis due to its highly aggressive and metastatic behavior, with pulmonary metastases occurring in approximately 67% of cases, and a high rate of local recurrence [4-6]. Diagnosis is typically made at a mean age of around 43 years, with no clearly identified risk factors, in contrast to secondary breast angiosarcoma (SBA), which more commonly affects women around 73 years of age and is often associated with prior radiotherapy or chronic lymphedema [1,3-5]. The annual incidence of breast angiosarcoma is estimated at 4.6 cases per million, with approximately 1.5% occurring in men [1]. Reflecting its rarity, a recent systematic review by Zhu et al. identified only 1,888 reported cases worldwide [7].
Clinically, PAB usually presents as a painless but progressively enlarging breast mass, sometimes accompanied by a sensation of warmth and bluish skin discoloration, as observed in our case [1,3-5]. These atypical clinical features, combined with the extreme rarity of the disease, frequently lead to delayed diagnosis. Imaging may raise suspicion but cannot establish a definitive diagnosis, owing to the absence of pathognomonic radiological signs [8]. Mammography typically shows an ill-defined mass, occasionally lobulated or oval, with focal vessel wall thickening and no associated microcalcifications, reflecting the parenchymal rather than ductal origin of the lesion. Notably, several authors report that up to 33% of cases are initially interpreted as benign, underscoring the heterogeneity of imaging presentations. Ultrasound, particularly useful in dense breast tissue, often demonstrates increased vascularity; however, sonographic appearances also vary widely among patients [1,5,6].
Breast MRI is the preferred imaging modality, typically demonstrating low signal intensity on T1-weighted images and high signal intensity on T2-weighted images, with variable enhancement and washout patterns depending on tumor grade [5,6,9,10]. In young patients presenting with rapidly progressive breast masses, MRI should be considered the imaging technique of choice, particularly for large lesions, as it facilitates earlier diagnosis and more timely therapeutic intervention, which may ultimately improve clinical management and prognosis. PET-CT and CT are primarily used for staging purposes; however, definitive diagnosis relies on histopathological examination [3]. Fine-needle aspiration is limited by a high false-negative rate of approximately 37%, while core needle or macrobiopsy may be technically challenging because of the highly vascular nature of the tumor [1,3,8].
Histologically, PAB exhibits considerable variability, with tumor cells that may be pleomorphic, spindle-shaped, or epithelioid [3]. PAB is classified into three grades, ranging from well-differentiated (grade I) to highly aggressive (grade III), the latter often associated with focal necrosis or hemorrhage [4]. Multiple grades can coexist within the same tumor, with higher-grade areas (II–III) frequently concentrated in the tumor center, which can make detection in biopsy specimens challenging [1,3-5]. This histological heterogeneity complicates diagnosis and necessitates the use of vascular immunohistochemical markers, such as ERG, CD31, CD34, or FLI-1, which can confirm the vascular nature of the tumor when positive [7]. In contrast, secondary breast angiosarcoma (SBA) often exhibits MYC oncogene amplification, supporting the notion that PAB and SBA arise from distinct genetic origins [3,11]. These distinctions are critical not only for accurate differential diagnosis but also for guiding potential targeted therapies. Given the strong vascular component of these tumors, particularly in younger patients, anti-angiogenic agents such as bevacizumab (an anti-VEGF therapy) may represent a promising treatment strategy [7].
PAB carries a poor prognosis because of its high metastatic and infiltrative potential [1]. Survival outcomes are closely linked to tumor size and the adequacy of surgical resection margins [8]. Ghareeb et al. report 5-year disease-free survival rates of 76% for grade I, 70% for grade II, and only 15% for grade III tumors [4]. Although some aspects remain controversial, surgery is considered the primary treatment, with radical excision aiming for at least 5 mm clear margins to minimize the risk of local recurrence [12]. Axillary lymph node dissection is reserved for cases with clinically or radiologically evident nodal involvement [1,5,13].
The recurrence rate after surgery alone is estimated at 33%, highlighting that surgical excision may be insufficient as a standalone therapy [10]. The role of adjuvant treatments, including radiotherapy and chemotherapy, remains a matter of debate [1]. Radiotherapy can improve local control, particularly for high-grade tumors or cases with positive surgical margins [3,9,12]. Chemotherapy, especially regimens based on taxanes or anthracyclines, may provide benefit depending on factors such as tumor size (>5 cm), rapid growth, high grade, family history, or the presence of distant metastases, as observed in this case [1,7,12]. Immunotherapeutic approaches, including anti-PD-1 and anti-PD-L1 agents, show potential, especially in tumors with an inflamed microenvironment; however, their clinical utility in PAB has not yet been established [14,15].
Further prospective studies are required to establish definitive therapeutic guidelines for PAB [7]. In the future, genomic profiling and molecular biomarkers are expected to play a key role in identifying patients who may benefit from personalized, targeted therapies, thereby potentially improving clinical outcomes [4].
Conclusion
In summary, PAB is a rare, aggressive breast malignancy with a poor prognosis. Diagnosis relies entirely on histopathological analysis, as clinical and radiological findings are nonspecific. Clinicians should maintain a high index of suspicion for PAB in any apparently benign breast mass and promptly perform macrobiopsies. The current recommended treatment is simple mastectomy with clear surgical margins, without routine axillary lymph node sampling. The absence of standardized guidelines for adjuvant therapy underscores the need for prospective, randomized studies to optimize management, although the rarity and aggressiveness of PAB pose significant challenges to conducting such trials.
Disclosures
Author contributions
All authors contributed to the care and follow-up of the patient and revised and approved the final submitted manuscript.
Patient informed consent
Patient provided informed consent for the publication of the case report.