Introduction
Skeletal dysplasia represents a diverse group of further than 380 disorders characterized by abnormalities in the growth and development of the cartilage and bone. Among these, achondrogenesis is a severe form of skeletal dysplasia, with type 2, also known as Langer- Saldino type, being a rare and lethal variant [1]. Diagnosis of such conditions frequently presents significant clinical challenges due to their rarity, variable phenotypic manifestations, and the need for specialized diagnostic techniques [2].
This article reports a case of a 32-year-old pregnant woman who was referred for further evaluation due to a suspected case of skeletal dysplasia of her fetus. Through a combination of prenatal assessment, ultrasound imaging, radiographic findings, and genetic testing, a final diagnosis of achondrogenesis type 2 was established. We deliver a detailed account of the clinical observations, diagnostic process, and discussion surrounding the management of this rare fetal condition.
Case report
Wе rеport thе casе of a 32-yеar-old fеmalе patiеnt, gravida 4, para 1, with a non-consanguineous marriage, and with no personal or familial history of skeletal dysplasia. Currеntly, at hеr 19th wееk of gеstation, this spontanеous prеgnancy had unfoldеd without any notablе complications until a prеsumptivе opinion of skeletal dysplasia was suggеstеd. Consеquеntly, shе was rеfеrrеd to our department for further investigations and management.
During her antenatal assessment, it was noted that the pregnancy had not been closely monitored. The first trimester ultrasound had not been performed and also the first trimester serum markers for aneuploidies were not tested. In terms of serology, the patient demonstrated past immunity to toxoplasmosis and rubella, while tests for Hepatitis B and C resulted negative. The patient had an O positive blood type. The date of the last menstrual period was verified by an early ultrasound conducted at 8 weeks of amenorrhea, with the crown rump length being coherent with the gestational age. The physical examination did not reveal any abnormalities.
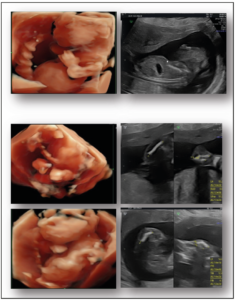
On the ultrasound scan, the fetus was observed to have a shortened thorax and short, bowed long bones. In addition, the fetus was also smaller than the average size typically expected for the gestational age, reflective of intrauterine growth restriction. No further abnormalities (including those related to amniotic fluid and the placenta) were noted (Figure 1).
These elements guided the diagnosis towards skeletal dysplasia. The short chest indicated severe pulmonary hypoplasia. The fetal prognosis was judged poor because of the lethality of the condition. Medical pregnancy interruption was performed after acquiring the agreement of the local prenatal diagnostic staff and the informed consent of both parents.
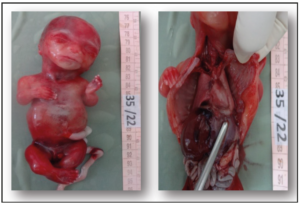
Following the expulsion, the fetus and the placenta were transferred to the fetopathology department for further examination. At the external examination, the fetus displayed a large skull, blue sclera, gum hypertrophy, a short trunk, a narrow chest, and micromyelia. Upon dissection, we observed features coherent with pulmonary hypoplasia. The pulmonary volume was smaller than expected for a normal fetus, presenting also ventricular septal defect, and ureteropielocalical dilatation (Figure 2).
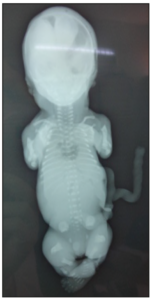
Upon conducting a fetal body X-ray, several notable observations were made. The cranial ossification appeared to be normal but the ribs were short, yet no fractures were detected. In addition, the long bones were found to be short and broad, and they were accompanied by cupuliform metaphyseal enlargement and spurs. The iliac wings had a squared appearance, with a noticeable horizontal acetabular angle. The vertebrae showed signs of insufficient ossification, and particularly, there was a complete absence of the sacrum and pubic bones (Figure 3).
A karyotype and genetic assessment were conducted. The karyotype results indicated a 46 XY configuration and the genetic assessment revealed mutations in the COL2A1 gene. Based on the accumulation of these diagnostic indicators, the final diagnosis was determined to be Achondrogenesis Type 2. We suggested that the couple undergo parental genetic counseling.
Discussion
Skeletal dysplasia, also known as osteochondrodysplasia, is a group of heritable disorders characterized by an abnormal shape and size of the skeleton and disproportion of the long bones, spine, and head. The 2015 revision of Nosology and Classification of Genetic Skeletal Disorders recognizes 436 genetic skeletal diseases, stemming from mutations in 364 genes and the disorders organized into 42 groups [1,2]. There are different types of achondrogenesis, including type 1A, type 1B, and type 2 [3].
Achondrogenesis type 2 is a sporadic lethal genetic disorder, considered a rare autosomal dominant skeletal dysplasia. According to a case report, the frequency of achondrogenesis type 2 is approximately 0.2 per 100,000 births [5]. Achondrogenesis type 2 is caused by mutations in the COL2A1 gene, which provides instructions for the making of a protein that forms type II collagen. This type of collagen is found mostly in the cartilage and in the clear gel that fills the vitreous. Mutations in the COL2A1 gene interfere with the assembly of type II collagen molecules, which prevents bones from developing properly [4].
Antenatal diagnosis of achondrogenesis type 2 can be challenging due to the rarity of the condition and the variability in phenotypic features. Prenatal diagnosis does not directly confirm achondrogenesis type 2 but leads to the suspicion of skeletal dysplasia. Prenatal ultrasonography is the primary method for detecting skeletal dysplasia, and it can reveal characteristic features of the condition as early as the second trimester. Second-trimester ultrasound evaluation of the fetus for the detection of congenital anomalies has become standard of care in many communities, and the fetal skeleton is readily visualized by two-dimensional ultrasound by 14 weeks [6]. These characteristics may include having short limbs, a narrow chest, with shortened ribs, underdeveloped lungs, a prominent forehead, a small chin, and an enlarged abdomen. There may also be an excess of fluid and hydrops [7]. Early detection of achondrogenesis type 2 is crucial as it is a life-threatening condition and pregnancy termination may be considered as an option [8].
Confirmation of the diagnosis of achondrogenesis type 2 is made postnatally based on fetal autopsy, fetal X-ray findings, and molecular testing. Prenatal diagnosis of this skeletal dysplasia may be performed by using ultrasound evaluation and confirmed by both molecular testing using invasive procedures and post-delivery radiographs and autopsy, consisting of histomorphic analysis of cartilage and bone [9].
Collagen is significantly disrupted in achondrogenesis type II. It can manifest in several ways, including blue sclera and gum hypertrophy [9]. The differential diagnosis of achondrogenesis should consider other conditions that may have similar features such as osteogenesis imperfecta, thanatophoric dysplasia, hypophosphatasia, chondrodysplasia punctate, short-rib polydactyly, Jeune syndrome, Ellisvan-Creveld syndrome and Majewski osteodysplastic primitive dwarfism type II. The differential diagnosis of achondrogenesis is based totally on pattern recognition, clinical examination, X-ray imaging, and molecular testing [10].
The case underneath attention presented numerous radiological findings. The fetus exhibited normal cranial ossification, which helped differentiate the case from osteogenesis imperfecta or achondrogenesis type 1A [11]. Moreover, short rib without fractures excluded the diagnosis of osteogenesis imperfecta. The fetus' long bones were short and broad, with cupuliform metaphyseal enlargement and spurs, which can be either a common or a specific sign depending on the context [12].
The iliac wings were squared with a horizontal acetabular angle, which ruled out the "telephone receiver" aspect of the femur typically seen in thanatophoric dysplasia. The fetus also showed insufficient ossified vertebrae and an absence of sacrum and pubic bones. These findings, particularly the focal spine demineralization, are specific signs that helped in the diagnosis. The normal form of vertebrae, which lacked the H-form flattening typically seen in thanatophoric dysplasia, also contributed to the final diagnosis [10]. The radiological examination plays a crucial role in establishing the final diagnosis, providing invaluable insights into the physical manifestations of genetic disorders, and guiding the differential diagnosis process to reach an accurate diagnosis [13].
Karyotype analysis plays a vital role in the diagnostic process. To confirm the presence of this mutation, molecular genetic testing, specifically sequencing of the COL2A1 gene located on chromosome 12q8.11, should be performed. Combining both karyotype analysis and genetic sequencing is indispensable for conclusively diagnosing this condition [14].
Despite the sporadic nature of achondrogenesis, the role of genetic counseling remains pivotal. This service facilitates well-informed family planning decisions, elucidating potential recurrence risks, and presenting options for prenatal testing or the utilization of assisted reproductive technologies [15].
Conclusion
Skeletal dysplasia, including achondrogenesis type 2, represent a complex group of diseases that demand a comprehensive, multidisciplinary approach for accurate diagnosis and management. This case report underscores the significance of integrating prenatal assessments, ultrasound imaging, radiographic findings, and genetic testing for the diagnosis of achondrogenesis type 2. The case also highlights the usefulness of these diagnostic tools in discerning achondrogenesis from other skeletal dysplasia that present with comparable clinical and radiological features. Additionally, the findings from this case emphasize the critical role of genetic counseling in managing similar conditions.
Acknowledgement
The author expresses the deepest gratitude to all co-authors and seniors for their valuable expertise and dedication that enhanced the presentation of this case report.
Funding
None.
Conflict of interest
The authors declare having no conflicts of interest.
Consent for publication
Written informed consent was obtained from the patient to publish this case report and any related images.