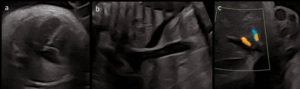Introduction
Noonan syndrome (NS) is a rare multisystem genetic disorder classified as a RASopathy, characterized by a variable phenotype that includes facial dysmorphism (i.e. low-set - posteriorly rotated ears, webbed neck), congenital heart defects, lymphatic and renal anomalies, and varying degrees of growth and cognitive delay. The incidence is estimated to be between 1 in 1,000 and 1 in 2,500 live births. Congenital heart defects are present in 50–80% of cases and include pulmonary stenosis, hypertrophic cardiomyopathy, ventricular and atrial septal defects, and Tetralogy of Fallot. Additional clinical features comprise: cryptorchidism, superior pectus carinatum and inferior pectus excavatum, coagulation disorders, ocular involvement, and an increased risk of developing childhood tumors [1,2].
The RASopathies are a family of disorders that include neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, capillary malformation–arteriovenous malformation syndrome, Costello syndrome, cardio-facio-cutaneous syndrome, and Legius syndrome. These conditions result from dysfunction of the intracellular Ras/MAPK signaling pathway, which is essential for proper cell cycle progression, growth, differentiation, and cellular senescence. Under normal conditions, this pathway is triggered by the binding of a growth factor to a receptor tyrosine kinase [3]. In RASopathies, mutations occur in genes encoding components or regulators of the Ras/MAPK pathway, leading to significant clinical consequences. Many phenotypic features overlap among these disorders, including facial dysmorphism, neurocognitive impairments, cardiac, cutaneous, musculoskeletal and ocular abnormalities, as well as an increased risk of malignancy [3].Most cases of NS are inherited in an autosomal dominant manner. The genes involved include BRAF, KRAS, MAP2K1, MRAS, NRAS, PTPN11, RAF1, RASA2, RIT1, RRAS2, SOS1, and SOS2. A rare autosomal recessive form is linked to mutations of the LZTR1 gene. Inheritance is from an affected parent in 30–75% of cases, while the remainder occur de novo. The most affected gene is PTPN11, mutated in approximately 50% of patients with NS [2].
PTPN11 encodes SHP2, a non-receptor protein tyrosine phosphatase with N-terminal and C-terminal SH2 domains and a catalytic PTP domain. Missense mutations in PTPN11 typically disrupt the autoinhibitory interactions between the N-SH2 domain and the PTP domain, resulting in inappropriate activation of the Ras/MAPK pathway, even without a growth factor bound to the tyrosine kinase receptor. The SH2 domains of SHP2 are extensively studied in oncogenetics due to their involvement in various cancers.2
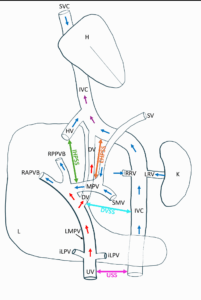
Normal fetal vascular anatomy includes three venous pathways (Figure 1):
- The systemic venous circulation, organized around the inferior and superior vena cava, which returns deoxygenated fetal blood from the periphery to the heart;
- The umbilical venous circulation, consisting of the umbilical vein (UV), joining the intrahepatic venous system;
- The portal venous circulation, with the main portal vein joining the UV within the liver, draining fetal blood collected via the superior mesenteric vein, inferior mesenteric vein, gastric vein, and splenic vein.
The ductus venosus (DV) is an intrahepatic venous connection linking the umbilical venous circulation to the systemic venous circulation by draining into the inferior vena cava (IVC) shortly before its entry into the right atrium. Three physiological fetal shunts normally exist: the DV, the ductus arteriosus, and the foramen ovale. These three shunts allow oxygenated blood from the chorionic villi to be redistributed into the systemic fetal circulation [4].
Agenesis of one of these physiological shunts, particularly the DV, has been reported. Agenesis of the DV is associated with the presence of an alternative shunt between the different fetal venous pathways [4], and these unusual connections are grouped under the entity of umbilical-portal-systemic venous shunts (UPSVS). Although the agenesis of the DV necessarily implies the existence of an alternative connection, the converse is not true: UPSVS can occur with or without associated agenesis of the DV.
According to Achiron et al. [5], UPSVS are prenatally classified into four types: type I (umbilical-systemic shunt [USS]), type II (ductus venosus-systemic shunt [DVSS]), type IIIa (intrahepatic portal-systemic shunt [IHPSS]), and type IIIb (extrahepatic portal-systemic shunt [EHPSS]) (Figure 1) [5].
In this document we report two cases from our Fetal Medicine Unit in which NS coexisted with type I UPSVS.
Case report 1
A 38-year-old G2P1 was referred to our Fetal Medicine Unit at 31 weeks of gestation due to fetal cardiomegaly secondary to DV agenesis, with the UV draining directly into the IVC. First-trimester ultrasound revealed increased nuchal translucency. Consequently, an amniocentesis was performed that showed a normal 46,XX karyotype. FISH analysis excluded a 22q11-microdeletion.
Ultrasound findings included:
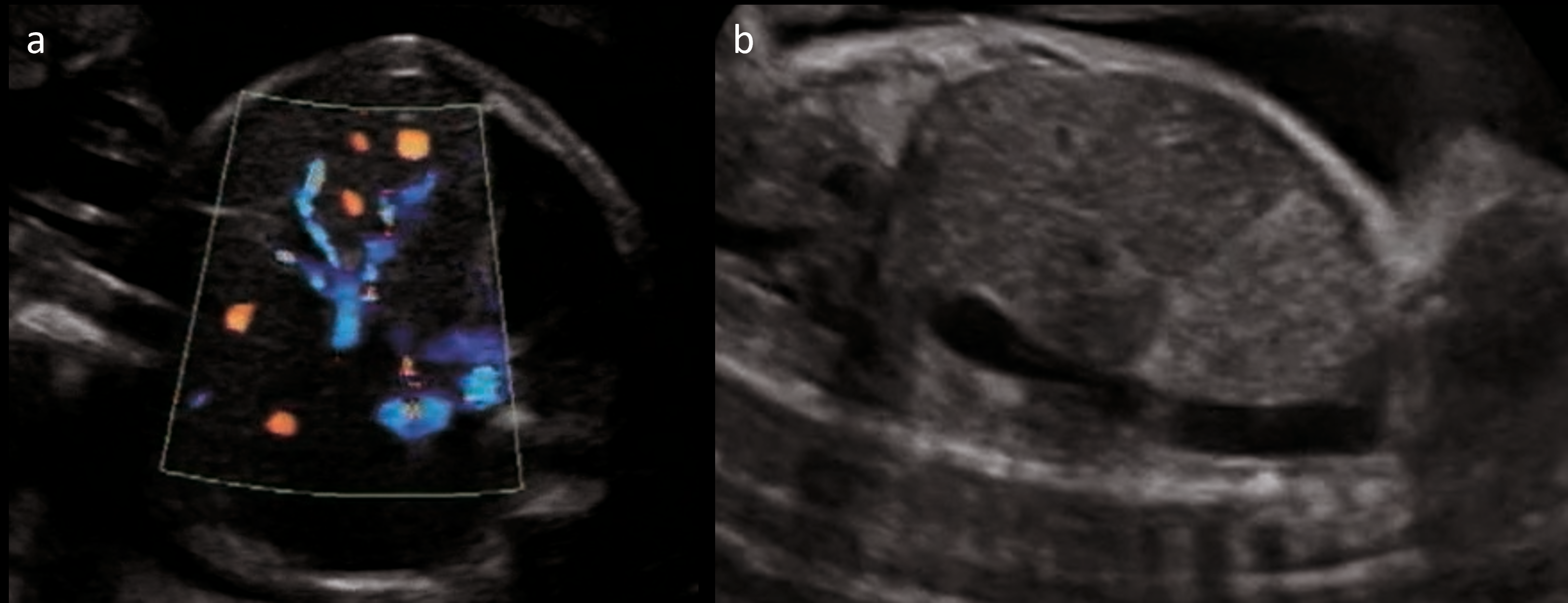
- DV agenesis (Figure 2) with UV draining into IVC (Type I UPSVS) (Figure 3);
- Cardiomegaly with preserved cardiac function;
- Small muscular ventricular septal defect (VSD).
The infant was delivered at 39 weeks (birth weight 2,725 g, appropriate for gestational age). Postnatal findings included:
- Cardiomegaly, spontaneous VSD closure within months, atrial septal defect surgically closed at age 5;
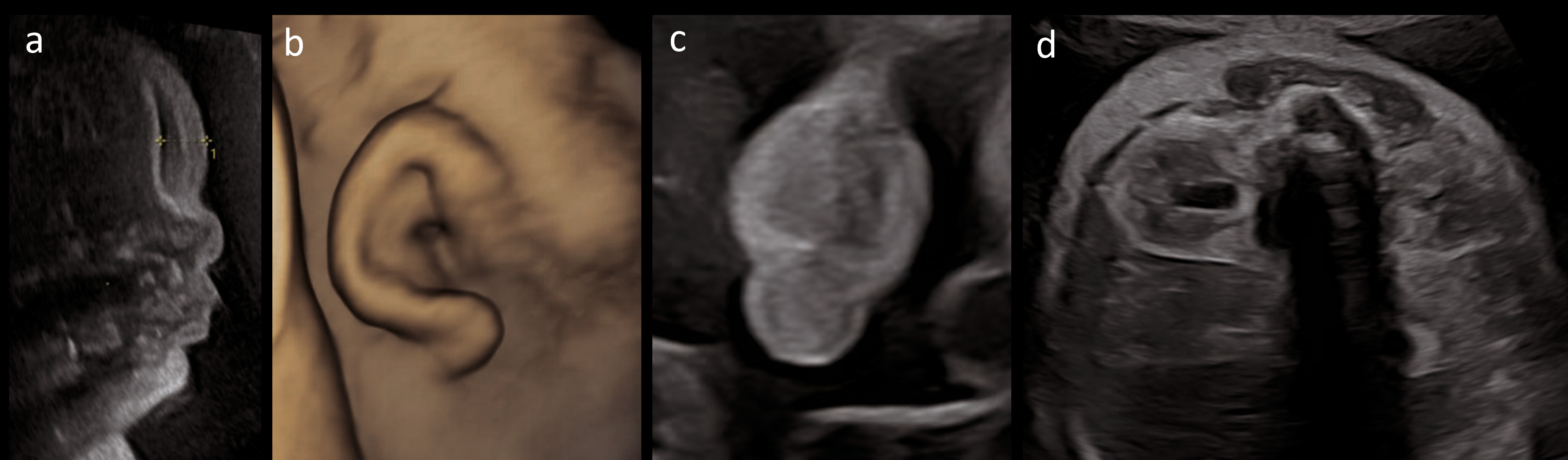
- Facial dysmorphism: cystic hygroma, hypertelorism, microcephaly, broad nasal bridge, long smooth philtrum;
- Widely spaced nipples;
- Persistent large anterior fontanelle; and
- Hepatic steatosis.
A RASopathy was suspected. Genetic testing confirmed PTPN11 mutation (c.179G>C p.G60A) [6]. Hemodynamic adaptation at birth was normal, apart from a transient need for supplemental oxygen for a few hours. Cardiac volume and function were normal on the first neonatal echocardiography. No vascular abnormalities were identified on postnatal abdominal ultrasound.
A diagnosis of mediastinal neuroblastoma was made at 5 months of age, for which the child was treated with chemotherapy and subsequently considered to be in complete remission.
Follow-up until the age of 12 years showed normal growth and satisfactory neurodevelopment.
Case report 2
A 39-year-old G5P3 was referred to our Fetal Medicine Unit at 29 weeks of gestation due to a polymalformative fetal presentation. First-trimester screening and non-invasive prenatal testing were normal.
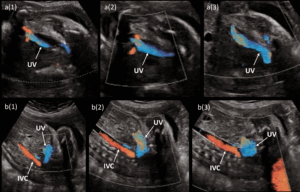
Ultrasound findings included (Figures 4 and 5):
- Severe polyhydramnios, requiring multiple amnioreductions;
- Frontal subcutaneous edema, nuchal thickening, and plantar edema;
- Macrosomia (estimated fetal weight and abdominal circumference > 99th percentile, Intergrowth-21st);
- Hepatosplenomegaly;
- Abnormal UV-IVC connection suggestive of USS, with early UV collateral and dilated IVC; normal DV Doppler;
- Small stomach;
- Echogenic right kidney with mild pyelectasis;
- Suspected bilateral cryptorchidism;
- Cardiomegaly with right ventricular hypertrophy and mild tricuspid regurgitation;
- Low-set, posteriorly rotated ears.
Molecular karyotyping was normal. Trio-based whole-exome sequencing revealed a de novo heterozygous PTPN11 variant (c.854T>C, p.F285S), consistent with NS and NS with multiple lentigines (LEOPARD syndrome, NSML). The pregnancy was continued.
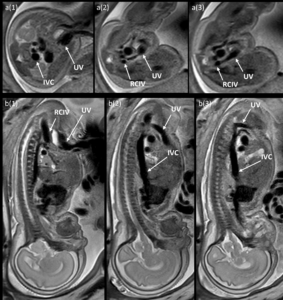
Fetal MRI confirmed a full-caliber UV draining into the right common iliac vein (RCIV), consistent with type I UPSVS (Figure 6). Subsequent ultrasounds showed fetal cardiac decompensation with right ventricular dilation/hypertrophy, pleural and pericardial effusions, and reversed a-wave in DV Doppler.
An emergency cesarean was performed at 32 weeks due to fetal distress. The neonate presented with generalized edema, petechiae, hematomas, dysmorphism, and died shortly after birth despite resuscitation. Birth weight was 3,345g (>99th percentile, Fenton 2013).
Discussion
These two cases illustrate the prenatal coexistence of NS, confirmed by exome sequencing, and type I UPSVS (USS).
A previous report from Sokollik et al. [7] have linked UPSVS with NS and NSML, though typically involving type III shunts.
Antenatal identification and classification of UPSVS are crucial for prognostic assessment and clinical decision-making. Type I UPSVS (USS) are poorly tolerated in utero, often resulting in hydrops, stillbirth, or premature delivery [8]. Achiron et al. [5] reported only 3 live births among 9 USS cases, with 2 early neonatal deaths and only 1 long-term survivor ("alive and well" survival rate: 11%) [5].
Following delivery, the closure of the UV often leads to a normalization of venous return and improvement in cardiac function. Postnatal imaging frequently fails to demonstrate the USS, emphasizing the importance of antenatal diagnosis.
Type I UPSVS are also notably associated with genetic anomalies (i.e. Down syndrome, Turner syndrome, RASopathy) [8]. Thus, identification of a USS prenatally should prompt detailed genetic evaluation including molecular karyotyping and targeted testing such as a RASopathy panel or trio-based exome sequencing – ideally following genetic counseling.
Other types of UPSVS have different prognoses: DVSS (Type II) usually has a favorable outcome, while Type III a/b may lead to postnatal complications such as thrombocytopenia or metabolic disorders potentially requiring intervention [5,8,9].
Multidisciplinary discussion is recommended for all UPSVS cases. While Types II and III may be managed at the referring center, type I UPSVS typically necessitates delivery at a tertiary care facility [8]. Jacquier et al. [8] advocate systematic screening for UPSVS during mid-trimester scans, using cranio-caudal abdominal sweep and sagittal abdominal views. Key features include a normal X-shaped portal sinus, IVC caliber, and hepatic penetration of the UV liver penetration (assessed by Doppler). Signs of cardiac strain (i.e. cardiomegaly, effusions, hydrops) warrant further evaluation.
Both of our cases presented with cardiomegaly. In one, tricuspid regurgitation and effusions were also observed. In both, the UV drained into a systemic vein. Variants with drainage into the right atrium or the coronary sinus have also been reported.
Menashe et al. [10] reported low antenatal detection rates of NS, especially in relation to cardiac anomalies. Prenatal diagnosis of NS is critical, even in milder cases, to optimize perinatal management and guide decisions including delivery planning or potential pregnancy termination.
Systematic screening for UPSVS and subtle signs of NS may improve prenatal detection. When such features are present, genetic counseling and comprehensive testing (molecular karyotyping, RASopathy panel, exome sequencing) should be offered.
In conclusion, although rarely reported, our cases suggest a noteworthy association between NS and Type I UPSVS. Recognition of this link may enhance prenatal detection of NS and improve perinatal outcomes through tailored management.
Patient consent
Written informed consent was obtained from patients for publication of their case reports and their accompanying images.