Introduction
Reed syndrome, or Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC), is a rare autosomal dominant disorder first described by Reed et al. in 1973 [1]. It is characterized by the presence of cutaneous piloleiomyomas and uterine leiomyomas in women, with a genetic basis linked to mutations in the fumarate hydratase (FH) gene located on chromosome 1q42.3–q43 [2]. This condition is associated with an increased risk of type 2 papillary renal cell carcinoma (PRCC) and, in rare cases, leiomyosarcoma [3]. Due to its rarity and variable presentation, the syndrome is often underdiagnosed. Although the overall incidence is low, early diagnosis of HLRCC is crucial due to its implications in oncologic risk and fertility. The consequences of a delayed diagnosis may include progression of renal malignancies and compromise of reproductive potential. This case report aims to raise awareness and emphasize the importance of early recognition and genetic screening.
Case report
A 35-year-old nulligravid woman presented in late 2023 with abnormal uterine bleeding and chronic pelvic pain. A transvaginal ultrasound performed in 2019 documented a large hypoechoic intramural mass measuring approximately 12 × 10 × 8 cm (≈ 2,000 cm³) and a uterine volume of 330 cm³. Magnetic resonance imaging (MRI) in 2020 confirmed uterine enlargement (15 × 15 × 11 cm) with associated pelvic varices.
Because of the substantial fibroid volume and marked vascularity, uterine artery embolization (UAE) was performed in 2022 as a fertility-sparing strategy prior to surgical management. Although UAE is not routinely recommended in young women desiring pregnancy, in this case it was considered due to the fibroid’s size and the anticipated technical complexity of primary myomectomy. Similar approaches have been described in selected reports of giant leiomyomas in young women [5,6]. Partial reduction of the mass was achieved, and the patient reaffirmed her desire for future fertility.
Histopathology showed a fusocelular pleomorphic neoplasm with a low mitotic index (<5 mitoses/10 high power fields). Immunohistochemistry revealed FH deficiency, consistent with leiomyoma associated with HLRCC. The report recommended genetic testing due to the risk of aggressive renal tumors. No histopathological images are available for documentation.
Follow-up transvaginal ultrasound on August 8, 2023, demonstrated a retroverted uterus measuring 7.0 × 6.5 × 6.0 cm (volume 143 cm³), with a pedunculated subserosal leiomyoma measuring 9.5 × 8.0 × 6.0 cm (volume ≈ 283 cm³). The endometrium was linear and uniform, measuring 8 mm in thickness. The right ovary was normal (6.5 cm³), while the left ovary contained a simple cyst measuring 63 × 68 × 58 mm (≈ 4.1 cm³).
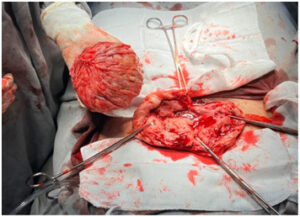
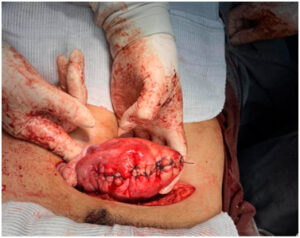
Given the persistence of symptoms and the patient’s reproductive desire, an open abdominal myomectomy was performed in December 2023. A single fibroid weighing 488 g, with areas of calcification, was excised. The uterus was reconstructed with layered suturing.
Histopathological evaluation revealed a spindle-cell pleomorphic neoplasm with a low mitotic index (<5 mitoses/10 high-power fields). Immunohistochemistry demonstrated loss of FH expression, consistent with FH-deficient leiomyoma associated with HLRCC. Diagnosis was therefore established on immunohistochemistry findings, as molecular testing was not performed.
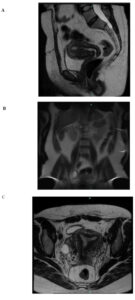
Family history was not available for this patient, which limited the possibility of assessing hereditary patterns within the family. The patient was referred to the oncology service for long-term surveillance. Abdominal and pelvic MRI in October 2024 showed no evidence of recurrence or postoperative complications. Expected postsurgical pelvic changes were observed, as well as a small periurethral cyst (1.2 cm) of no clinical relevance and a fat-containing umbilical hernia without incarceration or inflammation. Based on these findings, the plan was continued observation with the possibility of discharge from oncology follow-up if stability was maintained.
In 2025, the patient returned with complaints of infertility and is currently undergoing fertility investigation along with her partner.
Discussion
Reed syndrome, or HLRCC, presents with variable clinical manifestations ranging from multiple cutaneous leiomyomas to large, symptomatic uterine leiomyomas and, in some cases, highly aggressive renal tumors. The underlying molecular defect involves mutations in the FH gene, leading to loss of enzymatic activity and accumulation of fumarate, which functions as an oncometabolite. This promotes tumorigenesis through stabilization of hypoxia-inducible factors, epigenetic dysregulation, and metabolic reprogramming [2,3].
Up to 90% of women with HLRCC develop uterine leiomyomas, typically at a younger age and with tumors that are larger, more numerous, and more symptomatic than those seen in sporadic leiomyomas [4]. These tumors often grow rapidly and may be refractory to standard medical therapies, increasing the likelihood of requiring surgical management. Because FH-deficient leiomyomas may not display classic histologic features, targeted immunohistochemistry is critical for identifying FH loss. In this case, the diagnosis was established through immunohistochemistry demonstrating FH deficiency, even in the absence of confirmatory molecular testing [5].
The The present case gained further relevance as the patient returned to the gynecology service reporting infertility at age 37, prompting a comprehensive fertility evaluation. This underscores the long-term clinical implications of HLRCC and the importance of increasing awareness to promote early recognition and appropriate surveillance. As early as 2019, the patient already had a very large fibroid (≈12 cm, 2,000 cm³). Because of its considerable size and vascularization, UAE was selected as a bridging therapy to decrease tumor volume before surgery. Although UAE is not routinely recommended for young women desiring future fertility, isolated reports indicate that it may be considered in carefully selected cases of giant fibroids when primary myomectomy poses significant technical challenges [6]. Ultimately, an open myomectomy was required, resulting in the successful removal of a large calcified mass.
Another central consideration is oncologic surveillance. FH-deficient leiomyomas represent sentinel lesions for HLRCC, a condition associated with a substantial lifetime risk of developing highly aggressive type 2 papillary renal cell carcinoma. Current recommendations emphasize early, regular imaging surveillance [3,7,8], including annual abdominal MRI beginning in adolescence or at the time of diagnosis, due to the high metastatic potential of HLRCC-associated renal tumors [6,7]. In this case, the patient has undergone serial abdominal and pelvic MRI, all of which have shown no recurrence or evidence of renal involvement to date.
Beyond its oncologic implications, HLRCC may also influence reproductive health. Although a direct causal link between FH mutations and infertility has not been definitively established, factors such as the presence of large or multiple leiomyomas, prior UAE, and repeated pelvic surgery may adversely affect fertility potential. Additional contributors—including male factor infertility, diminished ovarian reserve, or other gynecologic or systemic conditions—should be assessed when evaluating reproductive outcomes in women with HLRCC. Theoretical mechanisms have also been proposed linking FH deficiency to impaired oocyte metabolism and ovarian reserve [8-10]. In this case, the patient’s infertility after myomectomy highlights the need for counseling regarding reproductive planning and consideration of fertility preservation strategies in women with early-onset or atypical leiomyomas [11].
Comparing this case with previously reported series reinforces its clinical relevance. While most published reports focus primarily on oncologic surveillance in HLRCC, far fewer highlight the reproductive consequences of the syndrome. The long-term follow-up of this patient demonstrates that HLRCC extends beyond cancer risk, affecting quality of life, fertility, and future reproductive planning. It also underscores the need for gynecologists to consider atypical, early-onset, or unusually large leiomyomas as possible indicators of HLRCC, prompting timely genetic evaluation and appropriate surveillance for patients and potentially affected family members.
Ultimately, increasing awareness among gynecologists, radiologists, and oncologists is essential, as early recognition of FH-deficient leiomyomas may serve as the first clue in identifying families at risk for this hereditary cancer syndrome.
Conclusion
Reed syndrome, or HLRCC, is a hereditary condition with significant gynecological, oncological, and reproductive implications. This case illustrates how early-onset and atypical uterine leiomyomas may represent a sentinel manifestation of the disorder. Prompt identification enables appropriate oncologic surveillance, ensures timely genetic counseling, and raises awareness of potential fertility consequences. Multidisciplinary management remains essential to optimize long-term outcomes for affected patients, highlighting the need for continued clinical vigilance and broader recognition of this entity in everyday practice.
Author contribution
Isadora Ferreira Kozlowski contributed to patient care and follow-up, conception of the case report, acquiring and interpreting the data, undertaking the literature review and critically revising the article for important intellectual content.
Marina Nunes Machado contributed to the initial diagnosis of the pathology, collaborated in the interpretation, and assisted in the writing of the manuscript.
All other authors contributed critically revising the article for important intellectual content.
All authors approved the final submitted manuscript.
Patient consent and ethics
Patient consent was obtained for the publication of this case report.
Ethical considerations were upheld throughout the evaluation of this case, publication that was authorized by the institution’s research and ethics committee. Measures were taken to ensure the confidentiality of information and patient anonymity.