Introduction
Inflammatory myofibroblastic tumors (IMTs) are rare and essentially benign mesenchymal tumors. They can be retroperitoneal, or found in the abdomen or pelvis (stomach, mesentery, omentum, urogenital tract), lungs, mediastinum, or head and neck [1]. The symptoms, which depend on the location and organs involved, are variable, for example vomiting, bleeding, palpable mass or intestinal obstruction. This great clinical variability and the lack of clear diagnostic imaging criteria make diagnosis of the condition difficult. Indeed, the diagnosis is often made only after surgical treatment. Management of the condition is not standardized, even though cases are preferentially managed in centers specializing in connective tissue tumors [2].
Case report
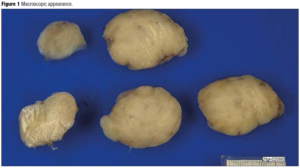
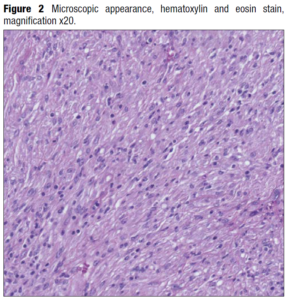
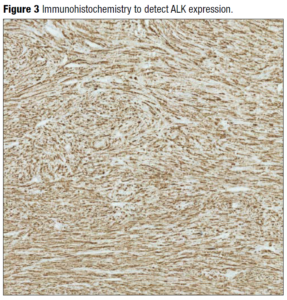
A 32-year-old woman presented for antenatal care in her third pregnancy. In her history we noted a first trimester surgical termination of pregnancy, and a forceps delivery at term which was complicated by a postpartum hemorrhage and episiotomybreakdown. The current pregnancy was a breech presentation, and the patient requested an elective caesarean section rather than external cephalic version as she had felt traumatized by her first delivery. The caesarean section was programmed for 38 weeks. During surgery, an oblong mass was felt in the left uterine horn (cornua). The mass was enucleated simply by sweeping the uterine cavity with a swab, without provoking any bleeding. The mass, which resembled a fibroid, was sent to histology for analysis. It had been situated next to the fetal head, which may explain why the fetus remained in breech position throughout the pregnancy, the mass possibly obstructing spontaneous version. The acoustic shadows of the cranial bones were probably responsible for the lack of detection at prenatal ultrasound screening. The histology results revealed an inflammatory myoblastic tumor. It was a well-defined, yellow nodular tumor measuring 40x35x25mm (Figure 1) with marked lymphocytic infiltration (Figure 2) and moderate anaplastic lymphoma kinase (ALK) expression (Figure 3). Estrogen receptors were poorly expressed, but progesterone receptors were significantly expressed and diffusely distributed. The mitotic index was low, at 5 mitoses observed in 10 fields at high magnification. Fluorescent in situ hybridization studies showed a rearrangement of the ALK gene at 2p23 in 60% of the nuclei analyzed. In view of the rarity of this tumor, the sample was sent to a specialist center in Bordeaux for confirmation. The case was presented at a multidisciplinary oncology meeting. The absence of any adjacent myometrium prevented adequate assessment of the margins of excision. The treatment plan proposed was to perform a sonogram of the uterus at the postpartum visit, followed by a magnetic resonance imaging (MRI) scan of the pelvis. In the event of recurrence, the positive ALK status would allow targeted treatment and follow up. The patient was seen at one month postpartum. There were no postpartum complications. The sonogram showed a normal uterus with no detectable nodule. A pelvic MRI was requested to complete the follow up, which was entirely normal.
Discussion
Inflammatory myofibroblastic tumors are mesenchymal lesions, which can be aggressive but usually behave in a benign manner. Their malignant potential is described as weak. The presence of IMTs in the uterus is described as rare, but probably underdiagnosed [3]; their etiology is a matter of debate, but some authors favor a traumatic or inflammatory origin. The prevalence of these uterine tumors is low, with 72 cases being described between 1987 and 2017 [4].
Diagnosis and histology
To help make the diagnosis, or to monitor the progression or regression of the tumor, the literature favors the CT scan with contrast, or the PET-CT scan. However, the MRI scan is favored in the pediatric population because of its excellent definition of soft tissues. Nevertheless, there are no strict diagnostic imaging criteria for this tumor. Histologically, IMTs have similar characteristics to smooth muscle, and are therefore easy to confuse with other tumors of the female genital tract, such as leiomyomas, leiomyosarcomas and endometrial stromal sarcomas [5]. In fact, IMTs are made up of a proliferation of polymorphic spindle cells and myofibroblasts on a stroma infiltrated by acute and chronic inflammatory cells, mainly lymphocytes. Nuclear atypia is frequent, and the mitotic index is in the order of 4.6 per 10 fields at high magnification. Classic smooth muscle markers, such as desmin, actin, vimentin, CD10 and caldesmone, are commonly detected [4, 6]. Detection of the highly specific [7] ALK gene rearrangement is essential to make a diagnosis of IMT and therefore to be able to offer specific treatment with the ALK inhibitors ceritinib and crizotinib, which can be lifesaving in cases of relapse or distant metastases. ALK protein expression occurs in approximately 50% of IMTs, but this increases to 88-100% in the case of uterine tumors [1]. This ALK gene activation is found in children especially. There would appear to be a strong association with pregnancy. In 2017, Pickett and his co-workers re-evaluated the diagnoses of a series of uterine tumors. Predictably, certain fibroids or sarcomas were redefined as IMTs. But the correlation with pregnancy was striking: 10.53% of the IMTs were discovered in pregnant patients as opposed to 0.17% in the non-gravid population (p=0.001). The majority of IMTs express estrogen receptors, and all express progesterone receptors. It is therefore logical to presume that these tumors would thrive in the favorable hormonal climate of the pregnant patient [5,8].
Management and prognosis
The prognosis is usually excellent. However, there is some debate over the behavior of the tumor. Some authors state that the prognosis is dependent on the size of the tumor and the presence of necrosis, while the resection margins and the mitotic index are unanimously recognized as prognostic factors. The gold standard in management is complete surgical resection. An expectant approach is also permitted in order to avoid major surgery or when seeking to preserve fertility. Occasional reassuring cases of spontaneous regression have also been reported in the literature [9]. Surgery should be minimally invasive, and equal success rates are reported with laparoscopy and hysteroscopy. Any fragmentation of the specimen should be avoided. Recurrence rates are low when the excision margins are clear but can reach 15 to 25% when resection is incomplete. Distant metastases are rarely encountered (2-5%), but when present they are usually found in the lungs, brain, liver or bones [4]. Given the low rates of metastases and recurrence under good surgical conditions, systematically aggressive surgery seems unnecessary. In these rare cases, crizotinib has been shown to be an effective therapeutic agent. This ALK inhibitor, which causes inhibition of cell proliferation, is recommended in cases where surgical resection cannot be envisaged, for distant metastatic tumors, or for recurrences [1]. In some case the treatment is associated with chemotherapy or radio-chemotherapy [10].
Conclusions
Uterine IMTs are rare but underdiagnosed, owing to their close resemblance to leiomyomas, as well as the fact that they share many histological markers. Only the presence of ALK expression allows accurate diagnosis. This same ALK expression is exploited by crizotinib, which has proved an effective therapeutic tool in appropriate cases. Given the strong association with pregnancy, gynecologists and obstetricians are encouraged to be aware of this tumor, which might be encountered during antenatal or peripartum examination. The prognosis is usually excellent, the tumor being benign in most cases, but a malignant potential is recognized. If the gold standard is surgical treatment, this must be qualitative, given the high rate of recurrence in cases of incomplete resection.