Introduction
Congenital lung malformations are historically rare clinical entities detected in approximately 1 in 10,000 to 35,000 live births [1], with an incidence that tends to increase with, current estimates ranging between 1 in 2,500 to 1 in 8,000 live births [2,3]. They include congenital pulmonary airway malformation (CPAM), previously known as congenital cystic adenomatoid malformation (CCAM), as well as bronchopulmonary sequestration (BPS), congenital lobar emphysema (CLE), bronchogenic cyst, and bronchial atresia. Hybrid lesions are common, suggesting that these rather represent a spectrum of abnormalities originating from a common pathogenic mechanism.
We report herein on a recent prenatal diagnosis of a large right pulmonary lesion with mediastinal shift. This clinical case illustrates that congenital pulmonary malformations represent a large spectrum of abnormalities, and that, despite an impressive prenatal diagnostic ultrasound, neonatal adaptation can, however, be favorable.
Case report
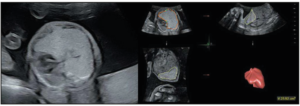
A 30-year-old woman in her first pregnancy, with a treated autoimmune hypothyroidism, was referred for morphological fetal ultrasound at 22+4 weeks of gestation (WG). The ultrasound revealed a large hyper echogenic pulmonary malformation on the right side of the fetus, with a volume of 25.9 cm3 causing a mediastinal shift without pleural effusion (Figure 1). There were no associated abnormalities, except for a velamentous insertion of the umbilical cord, nor were there any indirect signs of cardiac decompensation. Upon color Doppler ultrasound, the arterial supply of the mass could not be clearly identified.
The differential diagnosis was among a microcystic CPAM, BPS, and CLE. The CPAM volume ratio (CVR) at that time was estimated at 1.24, with a normal-high amniotic fluid volume, as reflected by an amniotic fluid index (AFI) of 17.8 cm.
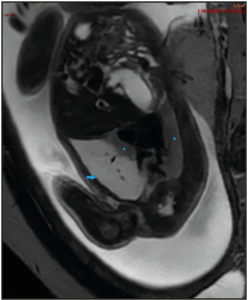
The patient was followed-up every week to monitor the risk of hydrops. The CVR increased to 1.61, along with the augmentation of the lesion’s volume to 36 cm3 at 24+4 WG, without any hydrops. The lesion turned out to be stable in volume from 26 WG to the delivery, though the mass was at the end smaller proportionally to the fetal size, while the mediastinal shift had decreased. A magnetic resonance imaging (MRI) was performed at 28 WA (Figure 2), confirming a right pulmonary lesion affecting almost the entire lung, with mass effect on adjacent structures. There were no systemic feeding vessels nor bronchocele identified.
Owing to the velamentous insertion of the umbilical cord, labour was induced at 39+3 WG using prostaglandins and oxytocin. Subsequently, patient gave vaginal birth to a girl of 3,165 g with perfect neonatal adaptation, displaying Apgar scores of 9/9/10.
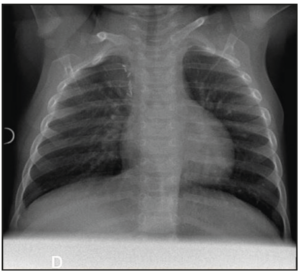
At day 1 of life, chest radiography and pleural ultrasound were carried out. The malformation was likely localized in the right upper pulmonary lobe, with a smaller volume than that antenatally detected, while the mediastinal shift was no longer present. The mass appeared solid, except for its anterior part where partial aeration was observed. There was an intern vascularization. Transthoracic echocardiography confirmed a normal cardiac anatomy and function.
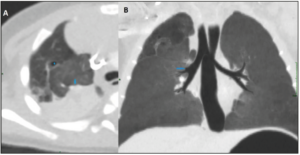
At 1 month of age, the COVID-19 virus infected the baby girl. With the exception of pyrexia, the girl stayed asymptomatic. A computed tomography (CT) was performed at 3 months of life (Figure 3), which further confirmed that an upper right lobe segment was not ventilated, while a stump of the upper lobar bronchus was identified. The diagnosis was in favour of bronchial atresia, with two additional antero-apical infra-centimetric (pseudo-) cyst formations identified.
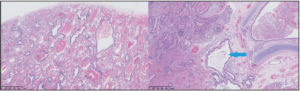
Following discussion with the parents and in order to prevent future risk of infection or malignant transformation, the decision was made to perform a surgical lesion resection. An open lobectomy was performed, with the girl being 4.5 months old. Upon the intervention, the upper segmental bronchus was filled with abundant mucus, without any aerial leakage at the opening. Pathological analysis revealed associated aspects of congenital CPAM type 2 with bronchial atresia. All these morphological findings were perfectly compatible with a hybrid lesion (Figure 4). Around 3 months after surgery, the chest radiography turned out to be totally normal (Figure 5).
Discussion
Congenital pulmonary airway malformation (CPAM), bronchopulmonary sequestration (BPS), congenital lobar emphysema (CLE), bronchogenic cyst, as well as bronchial atresia are well-accepted clinical entities. However, hybrid lesions are likewise common, suggesting that these masses rather represent a spectrum of abnormalities with the same etiopathogenesis [4].
CPAM is one of the most common lung lesions that are diagnosed prenatally (counter to CLE that is antenatally diagnosed in 25% of cases) [5], although its birth prevalence is rather low (1 in 10,000 live birth) [6]. A lack of clarity still exists in literature concerning its classification, pathogenesis, and management.
The first CPAM classification into three types was established by Stocker in 1977, based upon the size of cysts [7], which was further improved into five subtypes, with additional 0 and IV subtypes, in 2002. While this classification is quite useful for prognosis, it is not at all practical prenatally, owing to histologic analysis being required. The Adzick’s classification, established in 1985 [8], proposed, thus, two categories based on both gross anatomy and ultrasound findings. Macrocystic tumours contain single or multiple cysts of at least 5 mm in diameter, while microcystic tumours are likely more solid containing cysts less than 5 mm in diameter. This classification is, thus, easier to apply for in utero CPAM classification.
As our current comprehension of the pathogenesis of these malformations has evolved, revised nomenclature and classifications were developed to try to clarify the origin of these lesions and guide clinical management [9]. The Langston’s classification, established in 2003 [10], is based on pathologic features. A malformation sequence based on airway obstruction during development is proposed to unify the pathogenic mechanism of these overlapping lesions. We know that the CPAM occurs in early stage of the lung development, during the pseudoglandular or the early canalicular stage as we can already detect it on the second trimester ultrasound [11].
There are mainly two suppositions regarding the common origin of these lesions. The first one concerns a focal stop of normal lung development during the branching morphogenesis, affecting epithelial differentiation and/or crosstalk between the mesenchyme and epithelium. A molecular insult on factors regulating the airway formation could initiate this malformative process [11]. The other proposal concerns a bronchial stenosis [4,12]. This obstruction could be organic (with material) or functional (due to molecular insult on factors involved in the bronchial motricity) [11].
Functional obstruction could explain the discordances in the identification of anatomical obstacle in the study of Kreiger et al. [13]13 who didn’t find any pathologic features of bronchial atresia on their 23 cases of peri-partum fetal lung resections. The future histologic appearance of the distal lung will depend on the timing, level and degree of this events [9]. The etiology of congenital lung lesions could thus be a mixture of molecular abnormalities with obstructive incident [11]. Some factors implicated in this abnormal lung development have been reported such as overexpression of FGF-10 [14], or dysregulation of the Ras and PI3K-AKT-mTor signaling [15]. Further studies are still needed to identify the elements initiating the malformative process. Understanding which molecular factors initiate this improper development could give opportunities to explore pharmacologic solutions in severe cases [15]. Nevertheless, the prognosis is likely more dependent on the lesion size, including its consequences on adjacent structures, such as mediastinal shift, pulmonary hypoplasia, polyhydramnios, or cardiovascular compromise, in addition to development extent of the unaffected lung, and presence of other congenital abnormalities, than on the identification of the exact lesion type [16].
The presence of hydrops, mostly considered as the consequence of a large lesion, has shown to be the strongest prognostic factor. The survival rate without hydrops occurrence has been reported to be ≥ 95%. To predict the outcome in CPAM, the CVR was developed [17], representing a measure of tumour volume normalized for gestational age (mass volume/head circumference). If exceeding 1.6, the CVR was able to predict increased risk of hydrops. The authors also demonstrated that the CPAM volume plateaued at 26-28 WG. Once this plateau was reached, the fetus was no longer at risk of hydrops. Some regression in absolute CPAM size was occasionally seen, whereas the fall in CVR after 26 weeks was mostly caused by fetal growth [17], as we observed in our case.
The degree of hypoplasia of the remaining lung has been shown to be another essential prognostic factor. A prenatal MRI is useful to assess the residual lung volume. With its precise morphological evaluation, MRI can also be used for the differential diagnosis [3].
The prenatal management consists in frequent follow-ups with ultrasound imaging to monitor the risk of hydrops, along with CVR and MRI evaluation of residual lung volume. Hydrops, if associated with a poor heart function, was deemed to be associated with a high risk of mortality; therefore, an intervention should be considered for this case scenario [18]. Hence, attempting to make a histological diagnosis with prenatal imaging does not have great interest because of the overlapping features and hybrid lesions. The generic term “Congenital lung malformation” could be used antenatally with a good description of the malformation (macrocystic/microcystic/mixed/lesion’s volume and location/arterial supply) and its consequences on adjacent structures as far as the final histologic diagnosis doesn’t affect the prenatal management [19]. The postnatal management is primarily dependent on the newborn’s respiratory symptoms. Postnatal assessment can be performed using chest X-ray and CT. Characteristic CT findings of a combined lung lesion may be decisive for the postnatal management [20], but only the histological examination is what can definitively support the diagnosis. If the patient is symptomatic at birth, surgical resection is recommended [1].
In asymptomatic patients, CT evaluation should be carried out during the first 3 months of life [16]. For these patients, the surgical versus conservative approach is still a matter of debate, as no consensus has yet been reached regarding the optimal timing for surgery. The arguments in favour of elective and early resection consist of having a final histological diagnosis as well as preventing superimposed infection and the rare risk of malignant transformation. The surgical resection is usually performed after the neonatal period and before 12 months of age based on the potential compensatory lung growth, given that alveolar multiplication is decisive during the first two years of life [16].
However, the necessary prospective studies for surgical or conservative management are limited by the absence of a widely accepted practical classification system. Prospective follow-up studies of asymptomatic children managed by a conservative approach would be extremely informative and would allow to identify predictors of spontaneous good evolution [11]. In our clinical case, the surgical option was chosen, and an open lobectomy was easily performed at the girl’s age of 4.5 months without any complications. The baby girl is now 11 months old, and she is developing really well. Therefore, despite an impressive antenatal ultrasound depicting a pulmonary airway malformation with mediastinal shift, the outcome was favorable, as there was no development of hydrops antenatally.
Conclusion
Congenital lung malformations are clinical entities with an increasing incidence including CPAM, BPS, CLE, bronchogenic cyst, and bronchial atresia. There is still a lack of clarity in the literature regarding the nomenclature of these lesions, their pathogenesis, and their management. Hybrid lesions are rather common, suggesting that these masses represent a spectrum of abnormalities, secondary to obstruction rather than a differential diagnosis. In any case, the antenatal and postnatal care is the same regardless of the final diagnosis.
CPAM is the most common lung lesion detected prenatally. The antenatal prognosis is dependent on the mass size and its secondary effects, including hydrops among others, hypoplasia degree of the remaining lung, and associated abnormalities.
Postnatally, affected patients may either present respiratory distress in the newborn period or remain asymptomatic. Surgical resection is deemed curative but is a matter of debate in asymptomatic patients. The surgical intervention is usually performed before the patient is 12-months of age, owing to the potential compensatory lung growth. In the absence of surgery, recurrent pulmonary infections are among the most common complications. In addition, a weak association with malignancy has been reported. More studies are still needed to delve the molecular factors initiating the malformative process and to identify predictors of spontaneous good evolution in case of conservative management.
Conflicts of interest
The authors declare having no conflicts of interest.