Introduction
Aicardi-Goutières syndrome is a rare, genetic, early-onset and progressive encephalopathy [1,2]. This disorder results in severe intellectual and physical impairment with microcephaly, dystonia and spasticity [2-5]. Other clinical features include hepatosplenomegaly, chilblains or sterile pyrexias [1-6]. The main findings in neuroimaging are intracranial calcifications of the basal ganglia, white matter changes and cerebral atrophy [1-5,7-11]. Another characteristic is chronic lymphocytosis and increased concentrations of interferon-alpha (INF-α) in the cerebrospinal fluid [3-12].
In most cases, the syndrome appears either in the neonatal period with that progresses after a few months of an apparently normal development [2,6]. Prenatally, clinical features mimic the sequelae of a congenital TORCH infection [2,5,7-11] which must be excluded. Another condition to be considered in the differential diagnosis is pseudo-TORCH syndrome (Baraitser-Reardon syndrome) [2].
Hence, AGS is both phenotypically and genetically heterogeneous [2-3,6,10]. This syndrome is most frequently inherited in an autosomal recessive pattern, but it is also possibly inherited in an autosomal dominant manner and in addition, in a few instances, the disorder can result from specific de novo mutations [2,6]. At the present moment, there are 7 genes of which mutations are related to this condition: TREX1, RNASEH2B, RNASEH2C, RNASEH2A, SAMHD1, ADAR and IFIH [5,7,13]. Prenatal diagnosis of this entity is rare but it would be desirable for prognosis, management and future counselling purposes. We present an Aicardi-Goutières syndrome case with prenatally diagnosed in our Center.
Case report
Our patient was a Spanish 37-year-old pregnant woman, gravida 4, para 1. Both members of the couple had no personal background of interest. Routine maternal serologic screening evidenced immunity for rubella virus and negative results for toxoplasmosis, Hepatitis B and C, and HIV. Upon the routine first-trimester sonographic examination, ultrasound findings were normal. The combined estimated risk for trisomy 21 was 1:4105 and < 1:10000 for trisomies 13 and 18. Second-trimester ultrasound findings were also normal.
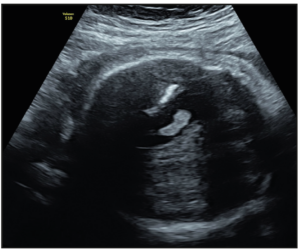
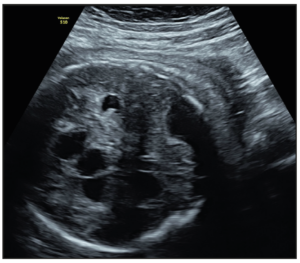
At the routine third-trimester sonographic examination (34 weeks of pregnancy), moderate bilateral ventriculomegaly was confirmed (15 mm at the atrial level) and other sonographic findings included an hyperechogenic periventricular halo and periventricular calcifications (Figure 1), enlarged cisterna magna (14 mm), and cerebral white matter hyperechogenic lesions like calcifications. The cerebellum was hypoplastic and it was displaced due to the enlargement of the cisterna magna (Figure 2). Hepatomegaly and cardiomegaly were observed. The estimated fetal weight was 2,200 gr (6th centile) with normal Doppler measurements.
Maternal serologic tests were performed, including cytomegalovirus (CMV), which was negative. Moreover, we performed an amniocentesis with the following results: fetal karyotype and CGH-array (KaryoNIM Prenatal®) were normal (46, XX), and the polymerase chain reaction for CMV, Listeria, Toxoplasma, Parvovirus, Hepatitis, Herpes, Zika and Chikungunya were all negative. Fetal magnetic resonance imaging was performed, confirming the ultrasound findings: tetraventricular hydrocephaly, intracranial calcifications, enlarged cisterna magna and hypoplastic cerebellum.
We additionally obtained amniotic fluid for exome sequencing of the fetus. A sequence variation of the RNASEH2B gene (c.476G>T) was found in homozygosis and a sequence variation of the ADAR gene (c.3002G>A) was found in heterozygosis. The missense variant in the RNASEH2B gene NM_024570.3:c.476G>T (p.Ser159Ile) rs76219783, which has no frequency data in population databases, is nevertheless classified as pathogenic in the HGMD database (CM074480), as it has been previously identified in a patient with AGS [6]. The missense variant in the ADAR gene NM_001111.5:c.3002G>A (p.Arg1001His) rs1571058184, has not been previously described and did not appear in the consulted databases. These pathogenic findings were associated with Aicardi-Goutières syndrome type 2 (AGS2, OMIM #610181) caused by homozygous or compound heterozygous mutation in the gene encoding subunit B of ribonuclease H2 (RNASEH2B).
At 37 weeks of gestation, our patient went into a labor induction because of intrauterine growth restriction (IUGR) and pathological Doppler measurements of umbilical and middle cerebral arteries. Cerebroplacental ratio was below the 5th centile and, for IUGR at this stage, our protocol recommends labor induction at 37 weeks following related guidelines. Cervical ripening was not indicated because Bishop score was over 7, hence, induction of labor with oxytocin was carried out with continuous fetal monitoring. There were no pathological findings on the fetal heart rate during all the induction. Finally, a 2,420 gr (5th centile) female infant was born by vaginal delivery. The Apgar score was 8-9-9 and umbilical cord pH were 7.21 and 7.34.
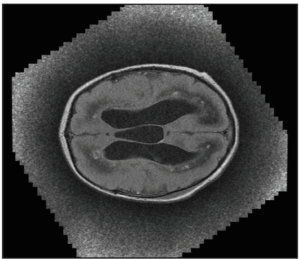
After birth, the pediatricians performed a transfontanellar ultrasound and a magnetic resonance and the findings were similar and confirmed those previously described (Figure 3).
Finally, we performed a genetic blood testing of both members of the couple and found the same sequence variation of the RNASEH2B gene (c.476G>T) in heterozygosis in both parents. In addition, we detected in the woman the same ADAR gene mutation, also in heterozygosis. Therefore, we concluded that both members were carriers of the Aicardi-Goutières syndrome type 2.
Currently, the infant is 3 years old. She is on rehabilitation and medical follow-up by pediatrics. She shows microcephaly, axial hypotonia with hypertonia and spasticity of the limbs. She suffers epilepsy, hypoacusia, dysphagia, respiratory failure and severe psychomotor retardation. These are a heterogeneous group of symptoms that are observed in patients with Aicardi-Goutières syndrome [3,4].
Discussion
In our case, we found a mutation in homozygosis in the RNASEH2B gene (c.476G>T) which is responsible of the disease (Aicardi-Goutières syndrome type 2). But moreover, both mother and daughter are heterozygous carriers of an undescribed but probably pathogenic mutation in the ADAR gene (c.3002G>A); although they did not have Dyschromatosis Symmetrica Hereditaria. the latter has only very rarely been reported outside of Japan and China.
To the best of our knowledge, there are only 5 cases of this syndrome published with prenatal diagnosis [9-11]. All of them had neurological impairment. The molecular findings were mutations in RNASEH2B gene in three cases and mutations in TREX1 in other two cases.
Aicardi-Goutières syndrome is a rare genetic and progressive encephalopathy that mimics a congenital infection, and it should be considered in cases in which one detects the described anomalies and a negative TORCH workup. Diagnosis is necessary for prognostic issues but also for the detection of parental carriers and proper prenatal counselling.
Author contributions
LDJ wrote the manuscript with support from RB. IO wrote the genetic segments of the paper and FC wrote the evolution of the child. FQ supervised the final text. All authors reviewed the draft paper and approved the final version of the manuscript.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.