Introduction
With improvement in ultrasound and molecular technology in prenatal diagnosis, more accurate fetal evaluation and genetic options are becoming available for the identification of rare monogenic disorders like Kabuki syndrome (KS). KS (OMIM # 147920) is a rare congenital polymalformative disorder occuring in 1/32,000 live births [1], caused by a pathogenic variant in the KMT2D gene (KS type 1), or less frequently in the chromosome X-located KDM6A gene (KS type 2). The lysin methyltransferase KMT2D epigenetically regulates the expression of developmental genes by histone methylation and thus, KMT2D loss impacts on broad aspects of the development [2]. Thirty percent of patients who clinically present as KS, do not have variant in either of these two KS genes, implying the existence of other known (RAP1A/RAP1B, HNRNPK, ZMIZ1) and unknown genes that may contribute to Kabuki-like phenotypes [3].
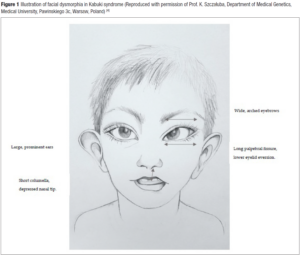
KS should be suspected postnatally in children with any combination of the five cardinal manifestations: skeletal and dermatoglyphic (persistent fingertip pads) anomalies, mild-to-moderate intellectual disability, growth deficiency and typical facial features [1]. Facial dysmorphism shows mainly long palpebral fissures with eversion of the lateral third of the lower eyelid, arched and broad eyebrows with the lateral third displaying sparseness or notching, a short columella with depressed nasal tip, and large prominent and/or cupped ears (Figure 1) [4]. Variable additional structural anomalies can be observed such as congenital heart defect (CHD), cleft lip and/or palate, gastrointestinal, genitourinary, ophtalmic or dental anomalies. Although prognosis is variable due to the multiple congenital anomalies and cognitive impairment that may lead to severe phenotype postnatally, the diagnosis is rarely done in utero [5].
The current case report describes the prenatal expression and the postnatal diagnosis of a KS. Thus, the present manuscript has aimed to 1) present a clinical KS case, with a confirmed de novo KMT2D pathogenic variant, admitted in the Fetal Medicine Unit of Saint-Luc University Hospital in Brussels Belgium; 2) review the prenatal characteristics of the syndrome; 3) discuss the contribution of prenatal dysmorphological facial analysis in this setting and 4) discuss the usefulness of WES in polymalformative syndromes.
Case report
A 30-year-old woman with no past medical history was referred to our tertiary center during the second trimester of gestation, because of asymmetric fetal cardiac ventricles and non visualization of the gallbladder. This pregnancy occurred 2 years after a first unremarkable pregnancy while partner remainded unchanged. There was no consumption of alcohol, tobacco, or any drugs during pregnancy. The first trimester of the pregnancy was uneventful, and no abnormality was detected by ultrasound, and the nuchal translucency was normal. The Non-Invasive Prenatal Test (NIPT) did not identify possible presence of aneuploidy in a probable male fetus. In our center, a first ultrasound was performed at 22nd week of gestation. We observed a fetal growth in the lower limits of normality (P25). Hypoplasia of the left heart ventricle and of the aortic arch associated with a possible aortic isthmic coarctation was diagnosed. The gallbladder was not seen at that time. The remaining morphologic assessment was normal and the amniotic fluid was within the normal range.
Presence of CHD entitled to offer invasive procedure to seek for submicroscopic pathogenic copy number variations (CNVs). QF-PCR ruled out trisomies 13, 18 and 21 as well as aneuploidy for the sex chromosomes. The molecular karyotype did not identify CNVs. In absence of the gallbladder, we performed quantification of specific enzymes (total proteins, alpha-fetoprotein and digestive enzyme activities including gamma-glutamyl transpeptidase) in the amniotic fluid as described by Dreux et al. [6]. Biochemical analysis revealed normal levels of digestive enzymes, excluding atresia of the bile ducts. Cystic fibrosis was excluded since no mutation of the CFTR gene in both parents was found (71of the most frequent mutations for the Belgian population) [7].
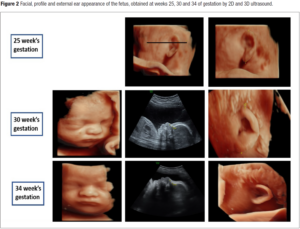
A first analysis of the facial morphology was attempted at the 25th week of gestation. Facial, profile and external ear appearance of the fetus obtained at weeks 25, 30 and 34 of gestation by 2D and 3D ultrasound are presented in Figure 2. No particular anomaly was observed at the 25th week of gestation. Ultrasound showed well-hemmed ears with a pavillion measured at 23.8 millimeters (P50). The gallbladder was still not visualized.
At the 30th week of gestation, amniotic fluid index was high (AFI 25 centimeters) thus revealing polyhydramnios. The estimated fetal weight remained in the lower normal limits (P20). Cardiac evaluation suspected the presence of a Shone’s complex, with a hypoplastic left ventricle, a hypoplastic aortic arch and a probable coarctation of the aorta. The mitral and aortic valves appeared non dysplastic. The evaluation of the facial features by our genetic team (Figure 2) revealed no distinctive facial dysmorphic signs. The ear was characterized as grade 2 microtia with absence of the crus of the helix. Orientation and position of the ear appeared normal and the pavillion was measured large at 30 millimeters (P93).
At the 34th week of gestation, a small for gestational age fetus (P9) was noted for the first time. The gallbladder was finally found. A new morphological analysis of the face was performed (Figure 2). Again, no special facial signs were identified, in particular the philtrum, the tip of the nose and palpebral slits were of normal appearance. Choanal atresia was ruled out because of the presence of a bilateral nasal flow with color doppler as well as facial asymetry. Grade 2 microtia, absent crus of the helix, protruding anthelix, squared superior portion of the helix, uplifted lobe and protruding ear were confirmed or identified. On the weekly multidisciplinary follow-up staff, a petrous temporal bone computed tomography (CT-Scan) to detect abnormalities associated for possible CHARGE syndrome (absence of semi-circular canals, hypoplastic canals, stenotic cochlear aperture) was offered, but the parents declined any further work-up because of the late age of the gestation.
The baby was born at 36 weeks and 3 days by vaginal delivery with an Apgar score of 9/10/10. Antibiotic prophylaxis was initiated because of premature rupture of membranes. The patient was immediately transferred to the Neonatal Intensive Care Unit because of the pathological cardiovascular context. Birth weight was 2,550 grams (P27), height 48 cm (P51), and head circumference 33 cm (P46), according to the Fenton charts [8]. The placenta was of normal constitution and weighed 304 grams (P5-10). Shone complex was confirmed by cardiac ultrasound (hypoplasia of the aortic arch associated with aortic isthmic stenosis and aortic bicuspid valve). Hepatic ultrasound was normal, and the gallbladder was seen. On Day 9 of life, aortic coarctation was successfully corrected. No additional dysmorphic signs were noted in the neonatal file.
At the age of 7 months, microcephaly (-2.5 SD), growth retardation (weight at -2.7 SD and length at -3 SD), gastroesophageal reflux and facial dysmorphism were recorded. Precisely, highly arched eyebrows, long palpebral fissures, short columella and prominent cupped ears were observed. The patient also had a high arched palate with a submucosal cleft and a bifid uvula (corrected by surgery at 14 months). The fingers were short with a clinodactyly of the fifth finger and fetal fingertip pads. This phenotype evoked in a first hypothesis as a KS. Since phenotype looked recognizable, targeted gene panel (SeqCap EZ, Roche, custom design) was selected and lead to the identification of the heterozygous c.11179del p.(Arg3727Valfs*22) truncating variant in the KMT2D gene (NM_003482). This specific variant has not yet been reported either in general population databases, such as GnomAD or ClinVar. Segregation study confirmed that the deletion arose de novo in the child since absent in DNA from both unaffected parents. Even if not reported yet, the present intragenic deletion was considered as pathogenic since it is responsible for premature stop codon as loss of function variants being a known mechanism leading to KS.
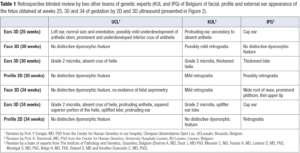
Facial, profile and external ear appearance of the fetus obtained at weeks 25, 30 and 34 of by 2D and 3D ultrasound presented in Figure 2 were retrospectively reviewed (with blinded diagnosis) by two other teams of genetic experts from our country. We wanted to evaluate the ability of other teams to identify any signs of possible facial dysmorphism on the same prenatal images. Their evaluations are presented and summarized in Table 1.
Discussion
KS is a rare and heterogeneous genetic disorder with reserved prognosis due to the combination of frequent multiple congenital anomalies. Even when fetal evaluation is performed by a trained sonographer and the pregnancy managed through a multidisciplinary collaboration, the diagnosis is rarely done prenatally as shown in our case report and in the literature [5].
Prenatal diagnosis of the syndrome is then challenging and few papers have been published on this topic. The postnatal diagnosis of KS is often made by the combination of developmental delay (intellectual disability, growth and motor delays, hearing loss and muscular hypotonia), multiple congenital anomalies and specific facial dysmorphism [9]. It is obviously impossible to evaluate prenatally the functional and neurodevelopmental aspects of the fetus. Nowadays, only structural malformations can be identified by prenatal ultrasound among these three clinical domains. Cardiac anomalies (49.4%), polyhydramnios (28.9%), genitourinary anomalies (26.5%), single umbilical artery (15.7%), intrauterine growth restriction (14.5%) or hydrops/pleural effusion/ascitis (12%) are the most common anomalies prenatally present in KS as reported by So et al. [5] in their case series published in 2021 combined with previous review of the literature. The anatomic types among patients with CHD are commonly left-side obstructive lesions and septal defects [9]. Shone complex with coarctation of the aorta was observed in our case such as a progressive polyhydramnios. Over a third of pregnancies in the study of Rosenberg et al. [10] were complicated by polyhydramnios possibly secondary to abnormal craniofacial structures and functional impairment of swallowing. Renal malformations include in order of prevalence: horseshoe kidney, renal hypodysplasia, renal ectopy or duplication. Urinary tract malformations include hydronephrosis and ureteral duplication [4]. Other sonographic anomalies can also be observed less frequently (<10%) such as an increase of nuchal translucency/cystic hygroma, central nervous system, skeletal or gastrointestinal anomalies [5]. More than one prenatal ultrasonographic abnormality were found in 69% of the cases reported by Rosenberg [10] thus reflecting the multi-organ involvement in KS. There is unfortunately no specific prenatal ultrasound feature with high sensitivity and high positive predictive value for KS diagnosis [11]. Malformations found in KS in the prenatal life indicate a phenotypic heterogeneity. Association of complex cardiac defects and renal structural anomalies was a common prenatal presentation in the case series of So et al. [5]. Differential diagnosis for KS with other conditions is often necessary. KS may mimic other disorders such as CHARGE syndrome, RASopathies, Cornelia De Lange syndrome, Turner syndrome, 22q11.2 deletion syndrome, oculo-auriculo-vertebral spectrum or VACTERL association. Other metabolic/infectious/immune/hematologic disorders may also show manifestations similar to hydrops [5,10].
Facial anomalies are found in 8.4% of the KS cases reported by So [5]. The most frequent are cleft palates. In our case, KS was diagnosed only postnatally in the presence of high arched palate with a submucosal cleft and a bifid uvula. There is no data about the possible evaluation of KS facial dysmorphism by 3D ultrasound. Our team has already reported in a previous publication a detailed analysis of the facial morphology (evaluation of forehead, eyes, palpebral fissures, nose, philtrum, mouth, lips, tongue, palate, chin, and ears) providing the clue to initiate molecular investigation targeted at a RASopathy [12]. In the present case, no facial anomalies were noted by the team neither prenatally nor at birth. A retrospective blinded evaluation of prenatal pictures of the face by two other teams of genetic experts of Belgium was not able to identify any significant prenatal dysmorphological facial signs that would have permitted to modify the course of the pregnancy (Table 1). As in other diseases but particularly in KS, the potential explanation of diagnostic failure is that subtle dysmorphic signs may not appear in fetal life but become obvious in childhood [13]. Moreover, despite careful examination, ultrasound evaluation faces technical problems: structures such as abnormal eyebrows are very difficult to investigate. Our team has also shown the feasibility and the interest in the evaluation of the external ears by 3D ultrasound (position, size and external appearance) to orientate diagnosis in the prenatal diagnosis of CHARGE syndrome [14]. In the present case, ear position was normal but both size and external appearance were abnormal. Anomalies (Grade 2 microtia, absent crus of helix, protruding anthelix, squared superior portion of the helix, uplifted lobe and protruding ear) were suspected at week 30th of gestation and confirmed at the 34th week of gestation with a P93 length of the pavilion. External ear dysmorphism in KS is nearly present in all patients (80-100%) and includes dysplasia, enlargement, external rotation, low-set or a cup shape. Rare reported anomalies have included microtia, preauricular fistula and pretragal tips [4].
After multidisciplinary discussion, a petrous temporal bone computed tomography (CT-Scan) to detect abnormalities associated with possible CHARGE syndrome (absence of semi-circular canals, hypoplastic canals, stenotic cochlear aperture) was offered, but the examination was declined by the parents because of the late age of the gestation.
In our case, due to late abnormal signs, no additional genetic evaluation was proposed after normal chromosomal micro-array. With advancement in prenatal molecular technologies and the discovery of new causative genes, KS can be diagnosed in prenatal genetic testing. In the literature review of So [5], the genetic diagnosis was established by single gene testing (N=17), targeted gene multiple panel (N=3), chromosomal microarray analysis (N=1), exome sequencing (N=36) and genome sequencing (N=1). But only 20% of the genetic testing was performed in the prenatal period and 80% were performed after delivery (after birth or postmortem). KMT2D pathogenic variants were identified in 91.6% and KDM6A pathogenic variants in 8.4%. Prenatal diagnosis of KS and appropriate genetic counselling must be improved and is essential for parents to be informed about the future health of the unborn child, hence, offering the choice of continuing and planning the childcare at birth or to terminate the pregnancy. Within the last 10 years, chromosomal micro-array has been routinely adopted to detect CNVs in prenatal diagnoses. Fetal structural anomalies can be associated with genetic disorders including aneuploidy, uniparental disomy, CNVs and intragenic mutations. There is increasing interest in genome-wide sequencing strategies to investigate prenatally detected congenital anomalies. Prenatal whole-genome sequencing has previously been described, but WES and targeted gene panels have received more interest because of their lower cost, the lower amount of fetal DNA required, a more rapid turnaround, and greater sequencing depth. The postnatal diagnosis was obtained in our case by a complementary genetic evaluation through a targeted gene panel at 7 months on the basis of expressed symptomatology at this age and because of the high probability of KS. Regarding the prenatal expression in our fetus, a WES evaluation would have probably been more adequate. This technique is available in our laboratories in Belgium and takes at least 3 weeks for final results (compatible with prenatal requirements). Several studies have shown a particular benefit of using this technique (in trio if possible) in a fetus with non-isolated congenital cardiac defects with normal micro-array [15,16]. A systematic review by Mone et al. [15] encompassing 636 CHDs revealed a significantly higher diagnostic rate of anomalies in cases of CHDs with multiple structural anomalies than in isolated CHDs (37% vs 11%). Li et al. [16] confirmed these results during the same time period (27.3% vs 7.9%). The most common genetic syndromes identified in the Mone study [15] were KS (19.8%), CHARGE syndrome (8.3%), Noonan syndrome (6.3%) and primary ciliar dyskinesia (6.3%). Finally, Sun et al. (17) showed incremental diagnostic yield of WES for single-gene defects in fetuses with congenital cardiac left-sided lesions (aortic valve atresia or stenosis, coarctation of the aorta, mitral atresia or stenosis and hypoplastic left heart syndrome) without aneuploidy or a pathogenic CNV. In his cohort, KMT2D was the most frequently mutated gene (7/66 [10.6%]) followed by NOTCH1 (4/66 [6.1%]). In this cohort, congenital cardiac left-sided lesions were the most prevalent cardiac phenotype (4/7) in cases with a KMT2D mutation.
In conclusion, prenatal diagnosis of KS remains challenging. In this document we report fetal case with a congenital cardiac left-sided lesion associated with poor growth, abnormal appearance of the external ear and polyhydramnios. This fetus was carrier for a novel KMT2D pathogenic variant compatible with KS. Although there is no specific ultrasound feature to easily recognize the syndrome, KS should be considered as potential diagnosis in fetuses with multiple congenital anomalies and particularly with cardiac left-sided lesions. The 3D-analysis of the face it-self was not helpful in our case, contrary to the external ear evaluation. In the future, a regular practice of WES in pregnancies with multiple congenital anomalies and heart defects will certainly increase in utero diagnosis of the syndrome offering parents the possibility to choose whether to continue or not the pregnancy or to plan the childcare at birth.
Declaration of generative AI and/or AI assisted technologies in the writing process
Authors did not use generative AI and/or AI-assisted technologies in the writing of the current manuscript.