Introduction
Steroid cell tumors are generally unilateral, benign and account for less than 0.1% of all ovarian tumors. Steroid cell tumors have 3 subtypes according to cellular origin: stromal luteoma arising from ovarian stroma, Leydig cell tumor arising from Leydig cells in the hilus, and steroid cell tumor (not otherwise specified, NOS) when the lineage is unknown. As far as ovarian steroid cell tumors, not otherwise specified (NOS) is concerned, it comprises about 56% of all steroid cell tumors [1]. The NOS subtype is highest in child bearing women, particularly during the third and fourth decades. This type of tumor can be functional and produce testosterone, leading to virilization, hyperandrogenism, and amenorrhea [2]. Around 56% of patients present with hirsutism. Steroid cell tumors have a wide variety of steroid secretions and estradiol is detectable in 6–23% of these patients [3]. Cushing’s syndrome has also been reported in 6% to 10% of patients [4]. It is worth noting that 25% to 45% of steroid cell tumors are clinically malignant [5]. Ovarian tumor markers are usually negative for steroid cell tumors [6]. On pelvic ultrasound, they are usually unilateral, solid, slightly hyper or hypoechoic lesions as compared to the ovary and not associated with ascites [7].
Case report
A 25-year-old year old young woman presents complaining of frequent menses, increased hair growth and acne for three months. She denied any voice changes, male pattern balding, mood changes, menorrhagia or dysmenorrhea. She had no history of contraceptive use, weight changes, stressors or significant personal or family medical history. On physical examination, there were no abdominal masses felt and there was no clitoromegaly noted and the Ferriman Gellway score was 8.
Investigations
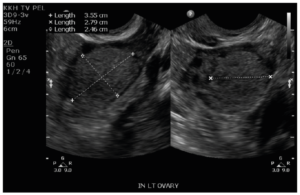
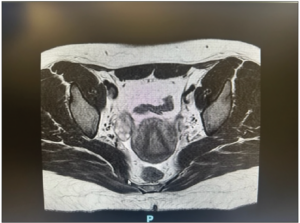
Laboratory analysis revealed raised total testosterone 6.0 nmol/L, free testosterone 0.086 nmol/L and raised 17 alpha-hydroxyprogesterone 24.8. DHEA-S and the short synacthen test was normal. Ovarian tumor markers CEA, Ca-125, beta-HCG and AFP were normal. Ultrasonography performed and repeated five months later revealed a stable 2.8 cm vascularized solid nodule in the left ovary with a normal uterus and right ovary (Figure 1). MRI of the pelvis showed a 2.5 cm T1 and T2 isointense lesion in the left ovary. It showed mild restricted diffusion and avid homogeneous enhancement with no fatty or calcific component within. There was no evidence of metastasis, ascites or enlarged lymph nodes (Figure 2). The right ovary, uterus, cervix and vagina were unremarkable. Initial endocrinologist impression was that of a benign ovarian tumor versus non-classical congenital adrenal hyperplasia or heterozygosity for 21 hydroxylase deficiency.
Treatment and follow-up
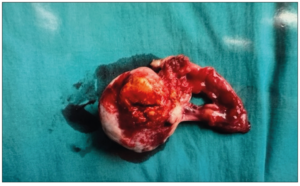
She underwent a diagnostic laparoscopy with left ovarian biopsy. Intra operative findings were that of a 4 cm yellow-white solid mass and the initial impression was that of a corpus luteal cyst. Final histopathology of the biopsy was worrisome for an ovarian steroid cell tumor. Multidisciplinary discussion was held and decision was for a total left oophorectomy. Six weeks later she subsequently underwent a laparoscopic left salpingo-oophorectomy. Intra operatively, it was noted that the right ovary and intra peritoneal look-up was normal. Final histology revealed steroid cell tumor of the left ovary with the tumor disrupted in the outer surface; peritoneal washings were negative. A gross specimen was received with the ovary measuring 45 x 40 x 20 mm. The outer surface of the ovary was mostly smooth and tan white but focally disrupted with tumor exposed to the surface. Cut section of the ovary showed a well circumscribed, solid, yellow nodule measuring 25 x 20 mm (Figure 3: Benign ovarian steroid cell tumor). Microscopic histopathology of the left ovary showed a circumscribed tumor with sheets of polygonal cells with clear vacuolated or eosinophilic cytoplasm and round nuclei. Mitoses were not readily identified (mean <1/10 high-power fields). No necrosis, hemorrhage, or marked atypia were observed. The overall morphology was diagnostic for a ovarian steroid cell tumor, of NOS type. The diagnosis was supported by the fact that her symptoms of virilization disappeared and menstrual cycle regulated after the lesion was removed. Repeat 17 alpha hydroxyprogesterone, free and total testosterone normalized two months after surgery. The patient has been on follow-up for five years with no evidence of tumoral recurrence.
Discussion and literature review
Steroid cell tumors are a rare subgroup of sex cord stromal tumors of the ovary. The incidence of this type of ovarian tumor is only 0.1% of all ovarian tumors. There are three subtypes of steroid cell tumors according to cellular origin: Leydig cell tumor arising from Leydig cells in the hilus, stromal luteoma arising from ovarian stroma and steroid cell tumors (not otherwise specified, NOS) when the lineage is unknown. As far as steroid cell tumors (NOS) is concerned, they constitute about 56% of all steroid cell tumors. This type of tumor can be functional and produce testosterone, leading to virilization, hyperandrogenism, and amenorrhea.
The most important factor to be determined in steroid cell tumors of the ovary is whether the tumor has malignant features or not. Hayes and Scully [8] identified five pathologic features for malignancy: two or more mitotic figures per 10 high-power fields (92% malignant), necrosis (86% malignant), size of 7 cm or larger (78% malignant), hemorrhage (77% malignant), and grade 2/3 nuclear atypia (64% malignant). Macroscopic examination usually shows a solid tumor; however, a combination of a solid and cystic form or predominantly cystic form may also be seen. The color of the cut surface may range from yellow to orange to red or brown depending upon the lipid content. Area of hemorrhage and necrosis may also be seen [9]. Microscopically, the NOS subtype of ovarian steroid cell tumor should be differentiated from a stromal luteoma and/or a Leydig cell tumor. Stromal leutoma is usually located in the ovarian stroma and there is presence of degenerative pseudovascular spaces containing red blood cells. Leydig cell tumors are usually present in hilar location and the tumor cells shows cytoplasmic reinke crystals and they are commonly associated with Leydig cell hyperplasia [10]. Most of the steroid cell tumors are positive for calretinin and inhibin, while negative for epithelial membrane antigen (EMA) in immunohistochemical staining [11]. Surgery is the most important and hallmark treatment for a benign steroid cell tumor, and complete excision of the tumor could provide the regression of symptoms and disappearance of the virilizing effects. The diagnosis of steroid cell tumors, NOS, should be made on the basis of the clinical virilizing signs, the microscopic and macroscopic pictures, as well as immune reactivity to some immunohistochemical markers. Therapy should be individualized based on tumor histology, surgical staging, and the desire for fertility preserving [12]. Malignant steroid cell tumors, NOS, should be managed with surgical removal followed by combination chemotherapy. BEP (bleomycin, etoposide, and cisplatin) regime is recommended to be an active first line chemotherapy regime for malignant stromal tumors [13]. Wang et al. [14] reported that the use of GnRH agonist as primary adjuvant therapy for sex cord stromal tumors including steroid cell tumors can be considered. Although steroid cell tumors are generally benign, there is a risk of malignant transformation and clinical malignant transformation. In young patients who are keen for fertility preservation, unilateral salpingo-oophorectomy is the preferred choice of treatment [15]. Steroid cell tumors in young women without evidence of malignancy on histopathology has excellent surgical outcomes and prognosis. In the present case, the patient presented with abnormal menstrual patterns, virilization signs as well as elevated testosterone and 17 alpha hydroxyprogesterone levels. A high index of suspicion of a steroid cell tumor should be maintained based in patients with a clinical presentation and blood tests similar to ours, and consideration for direct salpingo-oophorectomy be taken rather than firs an ovarian biopsy.
In conclusion, steroid cell tumors NOS are rare ovarian sex cord stromal tumors which can be difficult to diagnose. In a case of rapid-onset of hirsutism, virilization, and menstrual irregularity, it is extremely important to maintain a high index of suspicion. One can establish a final diagnosis according to the clinical manifestations and laboratory values in addition to imaging studies and laparoscopic examination of the ovarian mass. Malignancy is an important risk of steroid cell tumors, and pathologic evaluation is essential to rule out malignancy. Immunohistochemical testing is also helpful for the accurate diagnosis of a steroid cell tumor. Steroid cell tumors in young women without evidence of malignancy on histopathology have excellent surgical outcomes and prognosis.
Conflict of interest
The author declares having no conflict of interest.
Informed consent
Informed consent was obtained from the patient for the publication of this case report.
Financial disclosure or Funding
None.
Author contributions
The authors confirm sole responsibility for the following: collection of the information of the case and preparation of the document.