Introduction
Craniopharyngiomas are rare brain tumors that usually occur in children and young adults near the hypothalamus and pituitary gland. They are benign and typically do not metastasize to other sites of the brain or the body. Despite this, they can grow and exert pressure on nearby brain structures, including the pituitary gland, optic chiasm, and optic nerve, thus causing associated symptoms [1].
The degree of hypothalamic involvement can be classified by magnetic resonance imaging (MRI). In adults, visual field defects are the main manifestation at diagnosis, while these are detected later in children. Typical endocrinological deficits, secondary to pituitary compression, occur in over 60% of children but only in about 30% of adults. Short stature or delayed puberty are common manifestations of endocrine deficiencies in children. Adults mostly have more subtle hormone deficits, which might only be confirmed by blood tests [2,3].
Radical surgery offers the highest relapse-free survival and is indicated when hypothalamic integrity can be preserved. Craniopharyngioma treatment strategies have changed recently, varying from radical surgery (gross total resection or extended trans-sphenoidal endoscopic endonasal approach) to conservative surgery (sparing hypothalamus surgery) or radio-oncological techniques. Avoiding irreversible hypothalamic damage in pediatric patients is a key focus in the treatment of craniopharyngioma [4].
We report a complex case of primary amenorrhea secondary to craniopharyngioma surgery. In this patient, a panhypopituitarism developed following surgery, which furthermore led to multiple pathologies.
Case report
In September 2020, an 18-year-old female was referred to our clinic due to primary amenorrhea and a history of panhypopituitarism. In 2011, at the age of nine, the patient underwent a ventriculocisternostomy and subtotal excision of a craniopharyngioma and subsequently the placement of a ventriculoperitoneal shunt. Afterwards, she underwent radiotherapy.
Over the years, she developed panhypopituitarism, metabolic syndrome (severe obesity, fatty liver, insulin resistance, and hyperuricemia), long QT syndrome, and D hypovitaminosis. She was also found to be a carrier of HbS heterozygosity and AT III deficiency. No recurrence of the original resected tumor was registered at 10 years of follow-up.
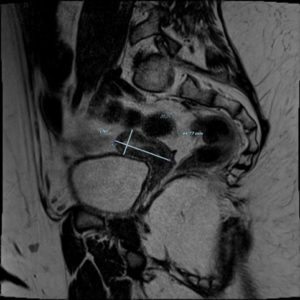
At the time of our medical examination, the patient was under treatment with hydrocortisone, vasopressin, levothyroxine, propanolol, cholecalciferol, metformin, and somatotropin. Furthermore, a transdermal patch of estradiol was started 2 years back (2018) and gradually increased to 25 mcg every 2-3 days. During our first evaluation, internal genitalia was impossible to visualize due to abdominal fat. Moreover, a pelvic MRI showed a small uterus for her age and ovaries with no functional signs consistent with a state of hypogonadotropic hypogonadism (Figure 1). The patient's mother reported the onset of thelarche on January 2020. The first assessment of pubertal development showed that her breasts were Tanner stage 2, and pubic hair was Tanner stage 1 (B2; PH1). After 3 years of estrogen therapy, partly sub-optimally dosed due to the patient's congenital thrombophilia, the Tanner score was B5; PH1.
In blood tests done in January 2021, the well-known hypogonadotropic hypogonadism (low estradiol, low LH, low FSH) was confirmed, which, together with the MRI results, suggested an under the dosage of the estradiol patch.
The patient's congenital thrombophilia and other risk factors made it difficult to tailor the satisfactory dosing of estrogen therapy, which was gradually increased but not yet adequately targeted to proper pubertal development of the patient. In February 2021, estrogen therapy was changed to a quadriphasic combined oral contraceptive containing estradiol valerate in combination with dienogest. At the last check-up, 4 months after the change of therapy, the Tanner's score was B5; estrogen levels were higher.
Discussion
Craniopharyngiomas are partly cystic benign epithelial tumors that originate from the squamous epithelial remnants of Rathke's pouch. The location of the tumor is determined by embryological events in the suprasellar region [5]. The overall incidence of craniopharyngiomas is 0.13-0.18 per 100,000 persons in adults and children. They constitute approximately 4-9% of pediatric brain tumors and 2-5% of adult intracranial tumors. The two major histopathological subtypes of craniopharyngioma are adamantinomatous and papillary [6,7].
Craniopharyngiomas can grow and compress nearby brain structures including the pituitary gland, optic chiasm, and optic nerve [1]. Surgical treatment is aimed at total resection while preserving hypothalamic integrity when possible. Prevention of irreversible hypothalamic damage is crucial, particularly important in pediatric patients and a key goal when treating craniopharyngiomas [4].
The presence of endocrine alterations is often one of the first signs of a craniopharyngioma; however, the onset of postoperative hypopituitarism is frequent and has been reported in several studies [8,9]. Patients can develop the necessity for multiple hormonal replacement therapies. In particular, in female patients, estrogen therapy may be needed to treat primary or secondary amenorrhea and delayed puberty [10,11].
Upon performing a literature search in PubMed using the words ‘amenorrhea and craniopharyngioma’, we found 17 articles. Of these 17 articles, 5 were not in the English language, 3 were not relevant to the search carried out, and 2 were not found in full-text.
Concerning the remaining articles: 4 were case reports, 2 were case series, and 1 was a review. In all described cases, amenorrhea (both primary and secondary) was reported among the signs present even before surgery [12-19].
None of the articles found addressed patient cases similar to the one we are reporting. Therefore, we extended the search to patients with amenorrhea and craniopharyngioma: 78 articles were found. Of these, 24 were not in the English language, 28 were not relevant to the search carried out, and 3 were not found in full text or abstract.
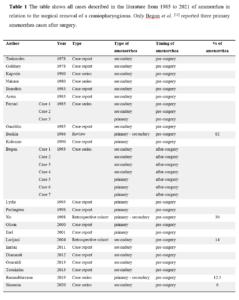
Of the remaining 23 articles, 14 were case reports, 6 were case series, 1 was a review and 2 were retrospective cohort studies [11-32]. In all but one described case, amenorrhea (both primary and secondary) was reported among the signs present even before surgery. In a case series of Begon et al. [12], there were 3 cases of primary amenorrhea after surgery. Two of these cases benefited from the administration of pulsatile GnRH. All cases are summarized in Table 1.
In adolescent girls, exogenous estrogen is crucial for the induction of secondary pubertal signs, growth, and bone mineralization [33]. A randomized controlled trial published in 2014 showed that transdermal estradiol resulted in higher estradiol blood levels and is more effective in breast and uterus development and feminization compared to orally conjugated estrogens [34].
In our case report, the panhypopituitarism developed after surgical removal of the craniopharyngioma, which necessitated multiple therapies due to hormonal deficiencies. In particular, estrogen therapy is necessary for pubertal development as the patient had primary amenorrhea with delayed puberty and breast development starting about 2 years after estrogen therapy inception.
The complexity of the clinical case is mainly a consequence of the patient's multiple pathologies that make satisfactory therapeutic dosing challenging to achieve. Upon our evaluation, the patient presented with severe obesity (body mass index [BMI] >40) with constant weight gain. In the adult population, several studies have identified obesity as a significant risk factor for deep vein thromboembolism (VTE). Obese individuals (BMI>30) have twice the risk of VTE than their normal-weight counterparts [35]. A study by Stokes et al. [36] established a correlation between obesity and VTE in a pediatric population, showing a similar risk of VTE in obese children to that described in adult cohorts [36]. Obese women who use oral contraceptives are more likely to experience VTE than obese women who do not. However, the absolute risk of VTE in healthy women of reproductive age remains low. The association of obesity with other common thrombotic risk factors can significantly increase the risk of VTE up to 23 times. Obesity is also an important factor related to the effectiveness of a hormonal contraceptive method. Some evidence, indeed, reports that the effectiveness of the patch has decreased among women who weighed more than 90 Kg [37].
Our patient also had AT deficiency. AT is a serine protease that inactivates thrombin and some clotting factors. AT activity is increased when it binds to heparin (both pharmacological and endothelial). Deficiencies in this, and other anticoagulant factors, are much more commonly acquired than inherited. Hereditary AT deficiency is a rare autosomal dominant thrombogenic disorder, with an estimated general population prevalence of 0.02-0.2%. Overall, AT deficiency carries a higher risk of developing VTE than the general population. Their relative risk of developing a first VTE is the highest of all inherited thrombophilias. In particular, the risk of thrombosis depends on the type of deficiency. Patients with a heparin binding site defect have the highest risk [38-40]. Estrogen is associated with numerous prothrombotic changes in the proteins involved in coagulation. First pass hepatic metabolism of oral estrogen leads to the increased hepatic synthesis of factor VII, factor X, and fibrinogen. Oral contraceptive users undergo several procoagulant changes in blood proteins, including increased levels of factors II, VII, VIII and X and fibrinogen, and reduced AT and protein S levels, and acquired activated protein C resistance [41,42].
QT prolongation can occur for many reasons, including electrolyte disturbances and myocarditis or secondary to drugs. Hypothyroidism is known to cause sinus bradycardia and delayed atrioventricular and intraventricular conduction, but changes in EKG repolarization such as prolongation of the QT interval and “torsades de pointes” due to altered ion channels are also recognized. Other hormone deficiencies may also be involved, for instance, hypogonadism has been associated with prolonging the QT interval, and cardiac arrest due to corticotropin and corticosteroid deficiency has been reported. Our patient underwent further investigations that reported a genetic mutation of uncertain significance regarding long QT syndrome [43-45].
In 2019, during a follow-up ultrasound examination, our patient was diagnosed with nonalcoholic fatty liver disease (NAFLD). NAFLD is the accumulation of macrovesicular fat in hepatocytes in the absence of excessive alcohol consumption or other known causes of fatty liver. Usually, it is not an isolated condition, but often a consequence of metabolic alterations resulting from high energy intake, obesity, sedentary lifestyle, and, above all, insulin resistance [46].
NAFLD is the most common chronic liver disease in adults and the pediatric population of the western world. A study by Mueller et al. [46] of more than 500 pediatric patients showed that low circulating levels of sex hormone binding globulin (SHBG), estrogen, and androgens were associated with more histologically severe NAFLD. Additionally, some animal model studies have reported a relationship between estrogen deficiency and increased susceptibility to NAFLD development, especially when associated with other predisposing factors such as a high-fat diet and a sedentary lifestyle [47]. In our patient, this condition was probably due to her lifestyle and obesity plus the hormonal alterations resulting from the surgery.
A satisfactory therapeutic dose was challenging to achieve due to the patient's multiple pathologies. The presence of severe obesity with insulin resistance, long QT syndrome, NAFLD, and hereditary AT deficiency caused a delay in the exogenous estrogen therapy replacement and its gradual increase. The change in therapy slightly improved the patient's pubertal development even if the therapy of choice in these cases remains the estrogen transdermal patch. This is likely due to the sub-optimal dose obtained with the transdermal estradiol patch therapy. Furthermore, the use of oral estradiol valerate at those dosages leads to higher plasmatic estradiol concentrations. However, the patient is continuing follow-up for her multiple gynecological and non-gynecological pathologies.
Conclusions
We report an interesting case of hypothalamic amenorrhea, panhypopituitarism, and multiple pathologies following the surgical removal of a craniopharyngioma at the age of nine. This case was challenging as the patient's multiple pathologies did not allow for a sufficient estrogen supply. In this case, estrogen therapy is essential for the patient's skeletal health and future fertility.
Acknowledgements: None
Conflicts of interest: The authors certify that there are no conflicts of interest with any financial organization regarding the material discussed in the manuscript.
Funding: The authors report no involvement by the sponsor in the presentation of this case.
Authors’ contributions: Author AT, VC, and FF have given substantial contributions to the conception and the design of the manuscript. Author AT and FF to acquisition of the data. All authors have participated to drafting the manuscript. Author LD revised it critically and finally all authors read and approved the final version of the manuscript.
Ethical approval: This case report involving human participants was compliant with the ethical standards of the institutional and national research committee and the 1964 Declaration of Helsinki and its subsequent amendments or comparable ethical standards. The case report was approved by the Institutional Review Board of DAME (Department of Medicine of the University of Udine).