Introduction
Placenta previa is a serious obstetrical complication associated with significant maternal and perinatal morbidity and mortality [1,2]; however, its management continues to be challenging [3]. Excessive or recurrent vaginal bleeding usually mandates delivery regardless of gestational age [4]. Vaginal bleeding due to placenta previa is associated with increased maternal morbidity, resulting in significant blood loss, the need for transfusion, and potentially postpartum hemorrhage (PPH), anemia, or cesarean hysterectomy, depending on the severity and location of the placenta [5]. Expectant management strategies vary according to clinical severity, yet there is no definitive method to alter placental location. Consequently, the therapeutic focus shifts toward reducing maternal blood loss and managing hemorrhagic complications [6,7]. Data on the use of interventional medications in placenta previa are limited and inconsistent, making their actual effect inconclusive [8].
Tranexamic acid (TXA) is an antifibrinolytic agent that has been shown to reduce blood loss and transfusion requirements in various medical, surgical, and gynecological conditions [9–12]. Furthermore, TXA has been widely used in obstetric settings [13–18]. Although TXA is classified as Category B during pregnancy [19] and has been investigated in other settings, significant uncertainty persists regarding its optimal use and dosage in cases of placenta previa. This clinical uncertainty necessitates formal randomized evaluation. We hypothesized that TXA could be an effective intervention to decrease the recurrence of vaginal bleeding and prolong pregnancy in women with placenta previa. Therefore, the current study aimed to determine the effectiveness of TXA in reducing vaginal blood loss and extending gestation, ultimately improving perinatal outcomes.
Methods
Study design and setting
This multicenter, randomized, double-blind clinical trial was conducted at five major public referral centers across Iraq and Egypt: Maternity Teaching Hospital in Erbil, Sulaimani Maternity Teaching Hospital, and Duhok Obstetrics and Gynecology Teaching Hospital, all located in the Kurdistan Region of Iraq; Azadi Teaching Hospital in Kirkuk, northern Iraq; and Al-Azhar University Hospital in Assiut, Egypt.
All participating sites are high-volume public teaching hospitals that operate 24 hours a day, 7 days a week, and serve as the primary referral centers for high-risk pregnancies and complicated obstetric cases in their regions. The annual delivery volumes at these institutions are exceptionally high. Based on administrative hospital records for 2023, Al-Azhar University Hospital reported approximately 12,000 deliveries, while the Maternity Teaching Hospital in Erbil recorded about 19,000. Delivery volumes at the other Iraqi hospitals are similarly high, with Duhok Obstetrics and Gynecology Teaching Hospital reporting 15,446 deliveries per year [20], and Sulaimani Maternity Teaching Hospital averaging approximately 16,000 deliveries annually [21].
Services at all five hospitals, including complex medical treatments and surgical procedures, are provided virtually free of charge to admitted patients. This policy effectively removes financial barriers to care, allowing access for women from all socioeconomic strata and attracting high-risk patients who require immediate and specialized management.
Participant recruitment, data collection, and intervention delivery for the clinical trial were conducted over a one-year period, from December 2022 to December 2023.
Eligibility criteria
After receiving independent approval from the relevant research ethics committees, 149 pregnant women with placenta previa were enrolled in the trial.
Inclusion criteria were as follows: pregnant women with confirmed placenta previa presenting with recurrent episodes of painless, minor vaginal bleeding sufficient to soil underwear or require a pad (estimated blood loss <50 mL). The bleeding had to be active (not settled or stopped) at the time of enrollment, and the mother had to be hemodynamically stable. Participants were required to be at or beyond 28 weeks’ gestation, regardless of parity, and to provide informed consent for participation.
Exclusion criteria included: placenta previa with accreta; presentation with major or massive hemorrhage (flooding or saturating multiple pads, with estimated blood loss >50 mL); severe abdominal pain; hemodynamic instability; fetal distress requiring immediate intervention; contraindications to TXA use (hypersensitivity, acquired defective color vision, or a history of venous thromboembolism); preexisting medical conditions that could affect pregnancy outcomes (i.e. diabetes mellitus, preeclampsia, heart disease, or renal disease); smoking; and refusal to participate in the trial.
Definition, diagnostic criteria, and grading classifications of placenta previa
Placenta previa was defined as a low-lying placenta located in the lower uterine segment [22]. Diagnosis was confirmed by ultrasound (US) performed by experienced sonographers at each hospital. Transabdominal US was performed first, followed by transvaginal US when indicated to more accurately determine placental location.
Placenta previa was categorized into three groups based on the distance of the placental edge from the internal cervical os:
- Marginal placenta previa: The distance between the placental edge and internal cervical os was less than 2 cm.
- Partial placenta previa: The lower edge of placenta partially overlapping the internal cervical os.
- Complete placenta previa: Placenta completely overlapping the internal cervical os [23,24].
In addition, ultrasound was used to assess the placental edge (including thickness and the presence of a marginal sinus) and to identify associated findings suggestive of Placenta Accreta Spectrum (PAS) [25]. Women with sonographic features suspicious for PAS were excluded from participation in the study.
Randomization and blinding
A total of 149 participants were randomly assigned in a near 1:1 ratio to one of two treatment groups: TXA or dextrose 5% in water. The allocation sequence was generated using block randomization with a block size of 4 via Random Allocation Software (version 2.0) [26]. This method ensured continuous balance (maintaining a 2:2 ratio after every four assignments) throughout the enrollment period, resulting in a final allocation of 75 participants in one group and 74 in the other.
After generating the random sequence, women were allocated to the two trial groups (TXA or dextrose 5% in water) using sequentially numbered, opaque containers with the corresponding treatment assignment. The preparation and numbering of these containers were conducted by an independent party not involved in participant recruitment or outcome assessment. At allocation, each participant was assigned a unique identification code (UI). This code was used during repeated hospital admissions, ensuring that each patient received the same designated intervention (either TXA or dextrose 5% in water) each time.
Identical 30-mL disposable syringes were used and were visually indistinguishable (same color, shape, and size), except for the unique UI labels, which concealed group allocation from the research team, including participants, caregivers, and outcome assessors. After completion of the trial, the UIs were decoded to determine each participant’s group assignment.
The sequentially numbered UI containers were secured in a designated, access-controlled area at each of the five hospitals and were dispensed sequentially upon enrollment of each new participant throughout the study period.
Source data for this trial were captured as part of each participant’s electronic clinical record at the respective center. Queries regarding participant data, adverse events, and overall trial progress were regularly discussed and resolved by the study leads (principal investigators) from each participating center to ensure consistent adherence to the protocol.
Sample size estimation
The sample size was estimated using PS computer software (Power and Sample Size Calculation, version 3.0.43). Due to the absence of published studies investigating the efficacy of TXA in reducing vaginal blood loss in placenta previa, an unpublished pilot study was conducted to obtain baseline data for the power analysis. This pilot study, which was independent of the main trial cohort, included two groups of 10 women each. Both groups followed the same study design and follow-up as planned for the main trial.
Based on the pilot study results, the following parameters were entered into the statistical power analysis for the primary outcome: a success rate of 90% for the TXA group and 70% for the control group. With a significance level (α) of 0.05 and a power of 85%, the estimated total sample size was 140 women (70 per group). To account for potential attrition and loss to follow-up, the final target sample size was increased to 149 women.
Grouping of the participants
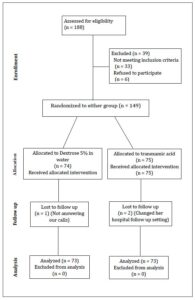
One hundred eighty-eight pregnant women with placenta previa were assessed for eligibility across the five hospital settings during the study period. Thirty-nine women were excluded because they did not meet the recruitment criteria or declined to participate. A total of 149 women were randomized to the two study arms. Seventy-five women were allocated to and received TXA, and 74 were allocated to and received placebo. Two participants were lost to follow-up and one was excluded from the analysis in the TXA group. One participant was lost from the placebo group. Ultimately, 73 women were analyzed in each group (as shown in Figure 1), which provides a flowchart outlining participant progression, trial design, and procedures in accordance with CONSORT recommendations [27].
Interventions
Tranexamic acid (TXA)
This intervention group received 1 g TXA (2 ampoules, each containing 500 mg/5 mL solution; Trenaxa; Macleods Pharmaceuticals Pvt. Ltd., Mumbai, India) diluted in 20 mL of Dextrose 5% in water, administered as a slow intravenous infusion twice daily for 48 hours during each episode of vaginal bleeding. This was accompanied by standard expectant management. A unique UI code was assigned for each hospital admission.
This dosing regimen was selected based on established pharmacological data. The 1 g IV bolus is the standard loading dose proven effective in the management of acute obstetrical hemorrhage [28] and trauma [29]. The twice-daily (12-hourly) frequency was chosen because the apparent elimination half-life of TXA is approximately 2 hours after IV administration, ensuring sustained therapeutic antifibrinolytic levels for continuous clot stabilization [30]. Furthermore, this intermittent, sustained regimen, with instructions to re-dose upon a new bleeding episode, directly addresses the known pathophysiology of placenta previa, which is characterized by recurrent vaginal bleeding.
Dextrose 5% group
The placebo group received 30 mL of Dextrose 5% in water administered as a slow intravenous infusion twice daily for 48 hours during each bleeding episode, along with standard expectant management. A unique UI code was assigned for each hospital admission.
Clinical trial outcome
Primary Outcomes:
- Sustained Hemostasis: Defined as the cessation of visible vaginal bleeding for a continuous period of at least 24 hours, with initial cessation noted at 4 hours. The study protocol mandated continuation of the intervention for a total of 48 hours.
- Pregnancy Reaching Term: Defined as continuation of the pregnancy after administration of the intervention, resulting in delivery at ≥37 weeks of gestation. This outcome served as a measure of the intervention’s success in prolonging pregnancy to full term.
- Favorable Perinatal Outcome: Defined as delivery of a newborn who met all four of the following criteria: (a) viable at birth; (b) birth weight ≥2,500 g; (c) Apgar score ≥7 at 5 minutes; and (d) no requirement for hospitalization or transfer to the neonatal intensive care unit (NICU) for medical management.
Secondary outcomes:
- Total Number of Antepartum Hospital Admissions: Recorded as the cumulative count of all distinct hospitalizations due to vaginal bleeding, pregnancy-related complications, or administration of the study intervention. This count included all admissions after enrollment up to, but not including, the final admission for delivery.
- Blood transfusion events (Inpatient and Outpatient): The proportion of participants who required transfusion of packed red blood cells (PRBCs) during the study period. The location of transfusion (inpatient or outpatient/day-case unit) was also recorded.
- Cumulative Units of Packed Red Blood Cells (PRBCs) Transfused: The total number of PRBC units administered to each participant, regardless of whether the transfusion occurred during an inpatient admission or on an outpatient basis.
Hospital Monitoring Strategies
managed under a standardized monitoring protocol across all trial centers to ensure the safety and efficacy of the intervention.
Vaginal blood loss was quantified throughout the admission using the gravimetric technique, which was the routine monitoring method for women with placenta previa. A calibrated digital scale was used at the bedside, and standard obstetric pads with known, pre-documented dry weights were employed for blood collection. Soiled materials were weighed at fixed 15-minute intervals during active bleeding or immediately upon saturation. Blood loss (mL) for each interval was calculated using the gravimetric principle, documented in the patient chart, and used to determine the bleeding rate. Women whose estimated blood loss exceeded the threshold defined for study inclusion (saturating or flooding underwear, or requiring multiple pads) were excluded and managed according to standard hospital protocols.
The primary endpoint was sustained hemostasis, defined as cessation of visible bleeding for at least 24 hours, with initial cessation assessed at 4 hours. To reduce recurrence, the study protocol required continuation of the assigned intervention for a total of 48 hours.
In addition to blood loss quantification, routine inpatient monitoring included frequent assessment of maternal vital signs, regular evaluation of fetal well-being, and monitoring of urine output to assess renal function. Specialist consultations (e.g., hematology or perinatology) were obtained when clinically indicated.
Women who responded to the intervention were scheduled for biweekly follow-up visits regardless of bleeding status, with more frequent visits arranged when required. Laboratory assessments included serial hemoglobin and hematocrit measurements during each admission, along with additional tests such as coagulation profiles when indicated. Blood transfusion was provided when clinically necessary. Antenatal corticosteroids were administered when preterm delivery was anticipated for maternal or fetal indications.
Scheduled delivery
The timing of cesarean delivery was determined according to gestational age, fetal well-being, and the severity of maternal bleeding. The clinical objective was to prolong pregnancy, when possible, to approximately 38–39 weeks of gestation. However, emergency cesarean sections were performed in cases of life-threatening maternal hemorrhage or non-reassuring fetal status.
All newborns received comprehensive pediatric monitoring from birth until discharge to ensure clinical stability prior to leaving the hospital.
Strategies for gathering data
Demographic data
Participant age, parity, and gestational age at admission were recorded. Gestational age was confirmed by first-trimester ultrasound at 11–14 weeks of gestation [31]. Body mass index (BMI) was calculated using the woman’s pre-pregnancy or early first-trimester weight and categorized as: normal weight (BMI 18.5–24.9 kg/m²), overweight (BMI 25–29.9 kg/m²), or obesity (BMI ≥30 kg/m²) [32].
Assessment of bleeding response
Cessation of vaginal bleeding was classified as either no response (bleeding did not stop) or gradual cessation following repeated intervention.
Hospital admissions and transfusion events
Cumulative hospital admissions were defined as hospitalizations requiring at least one overnight stay for monitoring active bleeding, managing complications, or administering the study protocol.
Blood transfusion events were recorded as any administration of packed red blood cells (PRBCs). These were often managed in outpatient or day-case settings to treat chronic anemia resulting from recurrent minor bleeding. Accordingly, transfusion requirements were assessed independently of inpatient admission status.
Criteria for blood transfusion
The need for transfusion was evaluated based on the cumulative effect of recurrent minor bleeding and the patient’s baseline hemoglobin levels. Decisions followed a standardized clinical protocol: transfusion was indicated for objective laboratory triggers (hemoglobin <7 g/dL) or for symptomatic anemia (e.g., persistent tachycardia or dizziness) resulting from depletion of physiological reserves. The total number of PRBC units transfused was recorded.
Mode of delivery
Delivery was categorized as elective cesarean section, emergency cesarean section, or vaginal delivery. A trial of vaginal labor was permitted only in cases of marginal placenta previa (placental edge >2 cm from the internal os), provided both maternal and fetal conditions were stable. This approach was applied to viable pregnancies deemed appropriate for trial of labor and to cases of macerated stillbirth.
Neonatal outcomes
Neonatal data included fetal viability, admission to the neonatal intensive care unit, and Apgar scores at one and five minutes. Scores were categorized as low (≤3), moderate (4–6), or reassuring (≥7) [33].
Gestational age at delivery
Gestational age at delivery was classified into four categories: very preterm (28–31+6 weeks), moderately to late preterm (32–36+6 weeks), early term (37–38+6 weeks), and full term (39–40+6 weeks) [34,35]. The discrepancy between gestational age at delivery and at initial admission was reported.
Birth weight classification
Birth weight was categorized as extremely low (<1,000 g), very low (1,000–1,499 g), low (1,500–2,499 g), or normal (2,500–3,999 g) [36,37].
Data management, safety monitoring, and adjudication
- Data collection and management methods
Trained research staff, blinded to the study purpose and unique participant identifier (UI) codes, collected initial data using validated paper-based Case Report Forms (CRFs) pre-coded with each participant’s UI. These forms were completed at the first participant interview according to the study protocol.
Data were subsequently transcribed from the paper CRFs into a secure, validated Electronic Data Capture (EDC) system for storage and analysis. The staff responsible for assessing the primary outcome were blinded to the participant’s treatment group assignment. All study data were accessible only to authorized personnel through role-based access control. The system was hosted on secure, compliant servers meeting relevant international standards for health data management.
An independent Data and Safety Monitoring Board (DSMB), composed of an obstetrician, a clinical trials methodologist, and an independent statistician, was established. The DSMB reviewed aggregate data every 14 days to monitor participant safety, adverse event rates, and the apparent efficacy of the intervention. An independent statistician prepared unblinded interim reports for the DSMB, operating separately from the core study team and the data entry process.
- Outcome adjudication
To standardize the final evaluation of primary and secondary outcomes, an independent Adjudication Committee was formed. The committee consisted of two external obstetrics and gynecology specialists with clinical trial experience who were blinded to treatment assignment. They reviewed individual participant data packages and applied predefined, standardized criteria to adjudicate and confirm all reported primary and secondary endpoints.
Adverse Event (AE) Reporting
Adverse Events (AEs) were defined as any untoward medical occurrence experienced by a patient receiving the study intervention (tranexamic acid), regardless of causality. A Serious Adverse Event (SAE) was defined according to regulatory standards as any AE resulting in death, a life-threatening condition, hospitalization, or persistent disability. Specific obstetric events, such as life-threatening maternal hemorrhage or non-reassuring fetal status leading to emergency delivery, were also classified as SAEs when they met the primary criteria for a life-threatening event or hospitalization related to the intervention.
All AEs and SAEs were documented on dedicated CRFs. Adverse events were reported to the sponsor within five calendar days of awareness. Non-fatal SAEs were reported to the sponsor, the Ethics Committee, and the DSMB within 24 hours of awareness. Fatal SAEs were reported to regulatory authorities within the same 24-hour period, followed by comprehensive reports within seven calendar days.
The independent statistician prepared unblinded summaries of aggregated AE data for the DSMB’s scheduled 14-day review meetings to maintain continuous safety monitoring throughout the trial.
Ethical approval
The research proposal received approval from the Research Ethics Committee, College of Medicine, Hawler Medical University (No. 9/10, September 11, 2022), covering the four research settings in Iraq. The Institutional Review Board of Al-Azhar-Assiut Faculty of Medicine (AZAST/RESEARCH/52/7-SEP-2022) approved the research setting in Egypt. Additionally, each local hospital’s institutional review board approved the proposal before participant recruitment. The study was conducted in accordance with the highest ethical standards, as outlined in the Declaration of Helsinki. Participants provided written informed consent after receiving comprehensive information about the study. Their right to withdraw from the study at any time without consequence was clearly communicated.
Statistical analysis
Data were analyzed using the Statistical Package for the Social Sciences (SPSS), version 26 (IBM Corporation, Chicago, IL, USA). Student’s t-test (unpaired t-test) was used to compare the means of the two study groups. The chi-square test of association was used to compare proportions. When the expected frequency in more than 20% of cells was less than 5, Fisher’s exact test was used instead of the chi-square test. Logistic regression analysis was performed when the dependent variable was binary (bleeding stopped vs. not stopped). Variables found to be significantly associated with the dependent variable in univariate analysis were entered into the regression model as independent variables. A p-value < 0.05 was considered statistically significant.
Results
Of the 149 women randomized, 73 in the TXA group and 73 in the 5% dextrose group completed the study and were included in the final analysis. Baseline characteristics were comparable between the two analyzed groups, confirming successful randomization, with no significant differences observed in age, gestational age at first hospital admission, or body mass index (BMI).
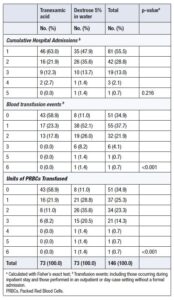
Notably, although the placebo group required significantly more transfusions, this did not result in an increase in total hospital admissions. This finding is consistent with the predefined secondary outcomes, which included transfusions administered in outpatient or day-case settings that did not require formal inpatient admission.
There were no significant differences between the two groups in overall inpatient hospital admissions. However, the placebo group had a significantly higher number of blood transfusion events (including both inpatient and outpatient/day-case events) compared with the TXA group, as shown in Table 1.
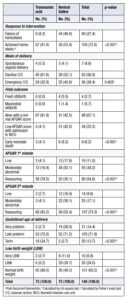
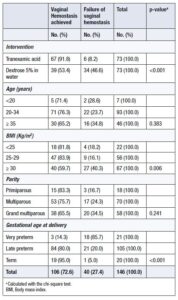
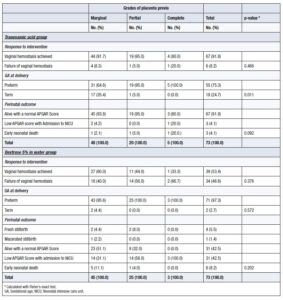
TXA demonstrated significantly greater efficacy than 5% dextrose in water in achieving vaginal hemostasis. Although most women in both groups underwent cesarean delivery, the TXA group was associated with significantly better neonatal outcomes, including higher rates of live births, normal-weight newborns, and normal Apgar scores. Furthermore, the TXA group had a significantly higher rate of term pregnancies (24.7%) compared with the placebo group, in which almost all deliveries (97.3%) were preterm (preterm rate in the TXA group: 75.3%) (Table 2).
Difference between gestational age at delivery and gestational age at first visit
The mean difference between gestational age at delivery and gestational age at first hospital admission was 5.6 weeks in the TXA group versus 1.7 weeks in the placebo group. Factors associated with cessation of minor vaginal bleeding in both groups were the use of TXA, BMI, and term pregnancy (Table 3).
In the TXA group, the grades of placenta previa were not significantly associated with bleeding cessation or overall perinatal outcome. However, there was a significant association between placenta previa grade and gestational age at delivery. In the Dextrose 5% in water (placebo) group, none of the outcomes—cessation of vaginal bleeding, gestational age at delivery, or perinatal outcome—showed a significant association with the grades of placenta previa (Table 4).
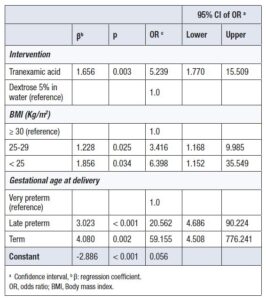
Factors significantly associated with vaginal hemostasis were the use of TXA (OR = 5.2; 95% CI: 1.7–15.5), BMI <25 kg/m² (OR = 6.3; 95% CI: 1.2–35.5), and BMI 25–29 kg/m² compared with BMI ≥30 kg/m², late preterm delivery (OR = 20.6; 95% CI: 4.6–90.2), and term delivery (OR = 4.5; 95% CI: 4.5–776.2) compared with very preterm deliveries (Table 5).
At the end of the study period, the TXA intervention group reported no adverse events or serious adverse events attributable to TXA administration, except for mild gastrointestinal symptoms (diarrhea and nausea) in three women. Specifically, no thromboembolic complications were recorded, including deep vein thrombosis, pulmonary embolism, myocardial infarction, or stroke.
Discussion
The study suggests that TXA may be more effective than placebo in managing vaginal bleeding associated with placenta previa. Although the data showed significant results, these findings primarily highlight the potential efficacy of TXA as a targeted intervention and warrant further investigation in this high-risk population.
The success of TXA in achieving cessation of vaginal bleeding after multiple doses can be directly attributed to its mechanism of action. As a synthetic lysine analog, TXA binds to plasminogen, thereby inhibiting fibrinolysis (the breakdown of blood clots) by preventing plasminogen from binding to fibrin. By preserving existing fibrin clots, TXA facilitates hemostasis and is widely used to reduce blood loss [38,39]. Clinically significant bleeding may result from surgery, trauma, obstetric complications, and various hemostatic disorders. Because the causes of bleeding are diverse and not always immediately apparent, the availability of a safe, effective, and nonspecific hemostatic agent is critical in many clinical settings, with antifibrinolytics commonly used for this purpose [10]. Likewise, as recurrent painless vaginal bleeding is the main symptom of placenta previa, treatment with TXA may help reduce vaginal blood loss.
The current study showed a higher rate of term pregnancies in the TXA group compared with the placebo group, as well as a longer interval between first admission, TXA administration, and delivery. It is well established that women with placenta previa presenting with antepartum bleeding have higher rates of preterm uterine contractions and preterm delivery [9,40].
The reduction in preterm birth rates was associated with improved perinatal outcomes in the TXA group compared with placebo. Preterm delivery is a common outcome in placenta previa and is associated with adverse neonatal consequences, including early neonatal death, NICU admission, and low birth weight [6,41,42].
The placebo group required significantly more PRBC transfusions and a higher total volume of units than the TXA group. Although inpatient admission rates were comparable between groups, the higher transfusion demand in the placebo arm was primarily managed through outpatient or day-case encounters, consistent with the study protocol. This finding aligns with a recent systematic review and meta-analysis of 50 randomized controlled trials showing that TXA reduced total blood loss and the need for transfusion in both low- and high-risk patients [43]. TXA is a well-established medication in obstetrics, with demonstrated efficacy in reducing blood loss, transfusion requirements, and bleeding-related mortality when used for PPH [44].
Most participants in this study underwent either elective or emergency cesarean delivery for placenta previa. This reflects the maternal and fetal risks associated with the condition, which often necessitate emergency cesarean delivery at any gestational age or planned cesarean delivery at around 38 weeks. Vaginal delivery was uncommon, occurring in 4.8% of cases, and was considered only in cases of marginal placenta previa with stable maternal and fetal conditions. Although this group included some macerated stillbirths, vaginal delivery was also successfully achieved in selected viable pregnancies where a trial of labor was considered safe. These findings underscore the importance of individualized management in placenta previa, considering not only gestational age but also maternal and fetal conditions, with a conservative, case-by-case risk–benefit approach.
In the TXA group, no significant association was found between the grades of placenta previa and vaginal bleeding outcomes. All types of placenta previa appeared to respond similarly to TXA in terms of reduced minor vaginal bleeding, prolonged pregnancy, and perinatal outcomes.
It is noteworthy that, in addition to TXA, normal or overweight BMI and late preterm to term delivery were significantly associated with cessation of vaginal bleeding in placenta previa. The potential relationship between BMI and bleeding risk is intriguing and warrants further investigation. Higher body size may influence bleeding tendencies in women with placenta previa. Brittany et al. reported that increased pre-pregnancy BMI is associated with greater maternal morbidity and bleeding among women with placenta previa. These findings highlight the importance of careful antenatal planning and peripartum management in this population [45].
The observation that cessation of vaginal bleeding was more likely in late-preterm births than in earlier preterm deliveries emphasizes the importance of gestational age in the clinical course of placenta previa and suggests potential mechanisms that merit further investigation in future studies.
Strengths of the study
The application of a randomized double-blind design played a role in controlling the effect of confounding variables. By including participants from different geographic locations and involving five teaching hospitals, we increased the generalizability of our findings to a broader patient population.
There is a wide gap in the literature concerning medical interventions for management strategies for minor vaginal bleeding during pregnancy among women with placenta previa. This study, therefore, is regarded as the first attempt to address this critical clinical issue. However, it is essential to acknowledge that, to generalize these results, larger-scale multicenter trials are warranted. Further research conducted in diverse healthcare settings will be crucial to validate our findings and improve management strategies for women with placenta previa experiencing minor vaginal bleeding.
A key strength of the study was the use of TXA, which is an easily available, inexpensive drug with a favorable safety record in obstetrics. Its use ensured that the study would be feasible to conduct and generalizable [14,15,20,46].
Limitations of the study
While the multicenter design is a strength, heterogeneity in patient characteristics and clinical practices across centers could influence the study outcomes. Larger studies with bigger sample sizes and broader enrollment areas are needed to further explore generalizability.
One of the limitations of this study was the absence of available data on maternal outcomes specific to placenta previa. However, this was not the primary aim of the study. Importantly, there were no cases of maternal morbidity or mortality in the study population. It is also noteworthy that women with hemodynamic instability and placenta accreta spectrum disorders associated with placenta previa were excluded, which likely reduced the risk of poor maternal outcomes.
Another limitation is the scarcity of literature regarding the efficacy of TXA for the management of placenta previa with minor vaginal bleeding during pregnancy. This restricts the availability of comparative data to place the current findings within a broader evidence-based context.
A critical limitation of this study was the selective inclusion criteria, which restricted enrollment to women with placenta previa experiencing only light and intermittent vaginal bleeding. Therefore, the positive findings regarding TXA cannot be generalized to all types of placenta previa, particularly those involving severe or massive antepartum hemorrhage.
Conclusion
This study offers initial evidence suggesting that TXA intervention in pregnant women with placenta previa experiencing minor vaginal bleeding may be a valuable therapeutic strategy. We observed a positive association between TXA administration and favorable gestational and perinatal outcomes. Further large-scale investigations with increased sample sizes are essential to confirm these findings, enhance generalizability, and better define the role of TXA in optimizing perinatal outcomes.
Level of Evidence: Level I; ClinicalTrials.gov: NCT05688111
Data availability statement
All raw, de-identified data, necessary to reproduce the findings of this study, including all versions related to the original and current analysis, have been deposited in the public repository ZENODO. The data are permanently and publicly accessible via the DOI: 10.5281/zenodo.17745387. This data deposit ensures full transparency and supports the corrected findings following the prior retraction of an earlier version of this study.